
Photoactivation-Based Labeling and In Vivo Tracking of RNA Molecules in the Nucleus
This protocol was adapted from “Photoactivation-Based Labeling and In Vivo Tracking of RNA Molecules in the Nucleus,” Chapter 11, in Live Cell Imaging: A Laboratory Manual (eds. Goldman and Spector). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA, 2005.INTRODUCTION
This protocol describes a method for observing and measuring the movement of RNA molecules in the nucleus of living mammalian cells. Caged fluorescein-labeled DNA oligonucleotides are introduced into living mammalian cells, where they demonstrably hybridize to complementary RNA. After site-specific photoactivation at desired sites within the cell, the RNA movements away from those sites are followed and digitally recorded using a rapid acquisition microscopy system.
MATERIALS
Reagents
Cells (plated onto 25-mm round coverslips, placed into 30-mm Petri dishes, and grown overnight to ~60% confluency)
Dimethyl sulfoxide (DMSO), anhydrous
Fluorescein, caged
The authors have exclusively used succinimidyl-ester-modified caged fluorochromes, which react well with amino-modified thymidines. Caged fluorescein succinimidyl esters show different rates of hydrolysis and aggregation in aqueous solution, depending on the chemistry of both the amino-reactive moieties and the caging groups. The compounds should therefore be stored desiccated, either as a solid or in anhydrous dimethylsulfoxide (DMSO) at -80°C, and suspended in an aqueous solution just before the coupling reaction. The caged compounds should also be protected from light.
Leibovitz’s L15 medium (GIBCO) containing 10% serum (no phenol red) or DMEM buffered with HEPES (no phenol red) (see Step 17)
Lipofectamine 2000 (GIBCO BRL)
In most studies using this method, oligonucleotides are complexed with a cationic lipid to facilitate their entry into cells. Note, however, that detectable levels will enter unaided if the oligonucleotide is added to the medium alone at an appropriate concentration (1 μM or more). The choice of cationic lipid is important, and the ratio of lipid to oligonucleotide must be optimized for each new oligonucleotide mix, following the manufacturer’s instructions for the particular cationic lipid in use. Currently, we use Lipofectamine 2000 (GIBCO BRL), but Tfx-50 (Promega) and Pfx-6 (discontinued when Invitrogen merged with GIBCO BRL) have also been used with excellent success. Oligofectamine (GIBCO BRL) gave low uptake of oligonucleotides in our initial trials.
Oligodeoxynucleotides
Oligodeoxynucleotides can be obtained from a commercial vendor (e.g., Integrated DNA Technologies) with amino-modified thymidines placed at predetermined positions in the sequence. The amino-modified thymidines should be positioned approximately every 10 nucleotides, partly to avoid intermolecular self-quenching of the fluorescein, but also, insofar as possible, to ensure that they are complementary to adenosines in the RNA target. In the case of an RNA target sequence that does not contain properly positioned adenosines, the modified thymidines may be placed at noncomplementary sites, but mismatches of more than two bases in the exact antisense sequence will decrease hybridization efficiency to an unacceptable level. The choice of the target RNA sequence and oligonucleotide design are important determinants of experimental success and should be given due attention. Target sequences most likely to be effective can be judged by several criteria, including the availability of an RNA site to the solvent environment (e.g., chemical mapping studies may have been carried out with the RNA of interest), the presence of protein-binding sites, and the RNA secondary structure (i.e., the ribonucleoprotein structure may be available, RNA footprinting studies may have been done, or the crystal structure of the RNA may have been solved). Two other key criteria in oligonucleotide design are the G+C content of the sequence (regions that are <50% G+C should be chosen) and its uniqueness (the chosen oligonucleotide cannot be one that has the potential to cross-hybridize with other cellular RNAs). Finally, it is important to use a control oligonucleotide in each experiment so that the movement of a free oligonucleotide is characterized under identical conditions.
Oligo(dT) or oligo(dA) 43-mer oligonucleotides or 33-mer anti-28S rRNA oligonucleotides (for sequence, see Politz et al. 2003) (see Step 12)
Opti-MEM (Optimized modified Eagle’s medium; GIBCO BRL)
Sodium bicarbonate buffer (1.0 M, pH 9.0)
Triethylammonium bicarbonate (10 mM, pH 8.5)
Equipment
Fluorimeter (optional; see Step 11)
Inverted epifluorescence microscope and imaging system (see Step 18) with heated chamber (see Step 15)
Lyophilizer or similar equipment (see Step 6)
Sephadex G-50 column
Spectrophotometer
TLC plate, fluor-impregnated (optional; see Step 7)
UV light box (e.g., FOTO/UV 300 Ultraviolet Transilluminator; Fotodyne) or hand-held UV lamp (e.g., model UVGL-58; UVP, Inc.)
METHOD
See Figure 1A for a diagram of caged fluorescein molecules. These molecules are coupled to oligonucleotides and then hybridized to RNA molecules of interest, as described below. The movements of the RNA molecules are visualized following photoactivation of the caged fluorescein-labeled oligonucleotides (see Fig. 1B for a cartoon of this strategy).
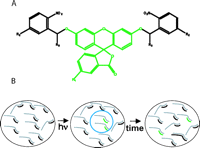
(A) Structure of caged fluorescein. R1, R2, and R3 indicate different groups in various caged fluoresceins that have been synthesized. The fluorescein ring system is locked into a nonfluorescent tautomer by virtue of attached o-nitrobenzene groups. (B) Cartoon showing uncaging strategy. The ether bonds that link the o-nitrobenzene groups to the fluorescein ring are photolabile. Irradiation with 360-nm light cleaves the ether bonds, and the fluorescein then rapidly adopts its fluorescent (xanthen-3-one) form. The black lines represent caged (nonfluorescent) oligonucleotides hybridized to RNA molecules (gray) inside the nucleus. The blue circle represents an uncaging site in which the green lines represent uncaged oligonucleotides hybridized to RNA. These molecules move out from the uncaging site over time (right side of diagram). (Modified from Politz 1999 and reprinted with permission from Elsevier © 1999.)
-
Coupling of Oligonucleotides with Caged Fluorescein
-
1. Dissolve oligonucleotides by adding a total of 100 nmol of modified amino groups (e.g., 25 nmol of oligonucleotide if there are four amino-modified thymidines per oligonucleotide) to ~130-140 μl of H2O.
-
2. Dissolve 1 mg of the caged fluorescein in anhydrous DMSO (usually 10-20 μl), and mix this with the amino group solution (prepared in Step 1) and 40 μl of sodium bicarbonate buffer (1.0 M, pH 9.0), to give a total volume of 200 μl. Allow the reaction to proceed for 12-24 hours at room temperature in the dark.
The water solubility of caged fluoresceins varies, thus if the compound precipitates, add more DMSO to the reaction mixture. The volume of DMSO used should not exceed one-third of the total volume.
-
3. Prepare a Sephadex G-50 column in a 25-ml pipette, and buffer it with triethylammonium bicarbonate (10 mM, pH 8.5). Load the reaction mixture onto the column.
-
4. Collect 1-ml fractions in microcentrifuge tubes, and then transfer 10-20-μl aliquots from each fraction to fresh microcentrifuge tubes.
-
5. Determine which of the fractions contain labeled oligonucleotide by uncaging and exciting the fluorescein: Stand the aliquots in an open-bottomed tube rack, and place the rack on a UV light box (~312-nm light) or use a hand-held long-wavelength (~366 nm) UV lamp. The presence of the oligonucleotide is indicated by green fluorescence, which may take several seconds to develop.
-
6. Lyophilize the oligonucleotide-containing fractions, and resuspend the labeled product in H2O (usually 100 μl).
-
7. Check the purity of the oligonucleotide by subjecting 2-3 μg to electrophoresis on a 10-12% (w/v) denaturing polyacrylamide gel (see Preparation of Denaturing Polyacrylamide Gels). Label the oligonucleotide of interest with standard fluorescein (as described here), and run it on the same gel as a marker.
The standard oligonucleotide will fluoresce immediately when the gel is placed on the UV box described in Step 5. The caged oligonucleotide should run as a single band (assuming a single oligonucleotide was labeled) that is initially nonfluorescent and then becomes fluorescent over time (seconds). If two bands (or more) are present with different uncaging characteristics, it is likely that the sample is contaminated with caged fluorescein aggregates of large size. If this is the case, the bands that contain DNA can be identified by placing the gel on a fluor-impregnated TLC plate and visualizing the dark (absorbing) DNA bands using a short-wavelength (~254 nm) UV hand-held lamp.
-
8. Aggregates must be removed or they will cause spurious results in live cell experiments. Remove contaminants by precipitating the oligonucleotide in 3 volumes of cold acetone (rather than ethanol) and 0.1 volume of ammonium acetate (3 M).
-
If this is not effective, purify the oligonucleotide by preparative gel electrophoresis (e.g., Purification of Synthetic Oligonucleotides by Polyacrylamide Gel Electrophoresis).
-
9. Uncage an aliquot of the purified oligonucleotide by transferring it to a microcentrifuge tube and lying it directly on the UV box for 30 minutes, which results in close-to-complete uncaging.
-
10. Determine the concentration of the oligonucleotide by measuring absorbance at 260 nm. This estimate will be high if the uncaging is incomplete. Determine the number of fluoresceins per molecule by reading the absorbance at 488 nm and then calculating the molar ratio of fluorescein to oligonucleotide (the extinction coefficient for fluorescein is 68,000 cm-1 M-1).
-
11. If necessary, determine the rate and degree of uncaging over time using a fluorimeter.
-
Introducing Caged-Fluorescein Oligonucleotides into Cells
-
12. Complex 4.5 μg of oligo(dT) or oligo(dA) 43-mer oligonucleotides or 9.75 μg of 33-mer anti-28S rRNA oligonucleotides with 6 μl of Lipofectamine 2000 according to the manufacturer’s instructions.
-
13. Add this complex to the cells (growing on coverslips in 30-mm Petri dishes) in a final volume of 1.5 ml of Opti-MEM.
-
14. Incubate the cells with the oligonucleotide for 2 hours and then in fresh medium (without oligonucleotide) for a further 30 minutes to 1 hour.
This treatment will facilitate efflux of internalized oligonucleotide that has not become bound within the cell.
Photoactivation and Tracking of RNAs
-
15. Mount cells growing on coverslips in a chamber suitable for use on a microscope stage.
We mount coverslips between two metal rings sealed with a gasket to form a chamber above the coverslip.
-
16. Heat the chamber (metal rings) with circulating water to maintain the cells at 37°C.
-
17. Use Leibovitz’s L15 medium containing 10% serum (no phenol red) or DMEM buffered with HEPES (no phenol red) at this stage of the experiment.
-
18. Mount the chamber on the stage of an inverted epifluorescence microscope (see Fig. 2) that incorporates a high-speed focus drive, wide-field laser illumination, and a small-format (128 × 128 pixels) frame-transfer CCD camera (MIT Lincoln Laboratory).
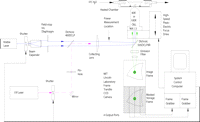 Figure 2.
Figure 2.Diagram of imaging system. All elements of the photolysis/imaging protocols (laser shutters, camera control, piezoelectric focus drive) are under the control of custom software running under Windows on a Pentium PC. The camera is thermoelectrically cooled, has ~70% quantum efficiency in the visible range, and has a readout noise of 6.5 electrons rms (root mean square).
-
19. Identify the nucleus to be uncaged in bright field (or use a phase-contrast lens and condenser), and move the region to be uncaged to the center of the field using standard stage controls. If diffusion times are to be measured, it is helpful to choose a site near the center of the nucleus so that the distance the signal travels before encountering (and being slowed by) the nuclear membrane is maximal.
Changes in cell morphology, especially retraction of lamellipodia, are an indication that the cells are being affected by the light used in imaging, and a new coverslip should be mounted. It is usually possible to image cells on a coverslip for ~2 hours without obvious detriment to cell health.
-
20. Uncage the signal.
The caged fluorescein is uncaged by the 351- and 364-nm lines of an argon laser (Coherent). The unexpanded UV beam, shuttered by a second LS3, is incident on a pinhole (Edmund Scientific, 35-μm diameter for 40X objective, 100-μm diameter for 100X objective) and combined with a 488-nm beam (see Fig. 2) by a 400DCLP dichroic mirror (Chroma, Inc.). The pinhole is mounted in a 3-axis positioner that allows it to be focused on the cell and centered in the field of the camera. To ensure consistency in our uncaging power, we measure (1815-C power meter, 818-UV detector; Newport Corp.) the UV power in the beam at a position between the collecting lens and the epifluorescence dichroic (see Fig. 2) several times during the course of an experiment. Uncaging is routinely carried out for 65 msec at a measured beam power of 90-120 μW (~1.6 kW/cm2 in the focused spot on the sample).
-
21. Collect and integrate fluorescence images of the resulting uncaged signal over time on the image frame of the CCD using exposure times ranging from 2 to 60 msec (operator controllable). An example is shown in Figure 3.
 Figure 3.
Figure 3.Movement of poly(A) RNA away from nuclear uncaging site. (Top) Distribution of uncaged oligo(dT) signal (bound to poly[A] RNA) in the L6 myoblast nucleus over time. The signal moves away from the site in all directions (except into the nucleolus) with an apparent diffusion coefficient of 0.6 μm2/sec. The bottom shows the much more rapid dispersion of the unhybridized control oligo(dA) in the L6 myoblast nucleus. Circles represent the approximate uncaging site.
Either 60 two-dimensional images (usually 10-msec exposure time with 500 msec between exposures) or five three-dimensional image stacks of 21 planes each (0.25-μm sections, 3-5 msec exposure time) are captured at 500-msec intervals after uncaging. The camera used to optimize this procedure was thermoelectrically cooled, had ~70% quantum efficiency in the visible range, and had a readout noise of 6.5 electrons rms. Uncaged fluorescein is excited by the 488-nm line of an argon-krypton laser (Coherent Inc.) that passes through a fast laser shutter (LS3; Vincent Assoc.), a beam expander, and a field-stop iris diaphragm (see Fig. 2). The beam then passes through a fused silica collecting lens, is reflected by a 505 DCLPXR dichroic mirror (Chroma Inc.), and is concentrated on the cell to be imaged by either a 40X NA 1.3 fluor objective or a 100X NA 1.3 fluor objective (Nikon). The optical configuration images the field stop onto the cell, so that the area illuminated can be limited to one slightly larger than a single cell. After the exposure is complete, the image is transferred to the CCD storage frame in 50 μsec and read out from four CCD output ports through four 2.5 Mpixel/second 12-bit analog to digital converters, and then to two frame-grabber boards, comprising a total of 8 Mb of dual ported memory (Bitflow, Inc.). The readout process is complete in 1.8 msec.
ACKNOWLEDGMENTS
The authors are deeply indebted to Timothy Mitchison for his generous gift of caged fluorescein and thoughtful discussions. Without his help, this line of research would have been very difficult to pursue. The collaborative role of Robert Singer during the early phases of this method’s development is warmly acknowledged, as is the key support of our late colleague Frederic Fay. This work was supported by National Institutes of Health grants GM-21595 and GM-60551; early stages of this work were supported by an NIH NRSA post-doctoral fellowship AR-08361 to J.C.R.P.








