
Assays with Protein Arrays
Adapted from “Characterization of Protein Complexes,” Chapter 10, in Proteins and Proteomics (ed. Simpson). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA, 2003.Protein-Protein Interactions
Protein arrays can be used to screen for protein-protein interactions. For this application, a protein microarray is incubated with a fluorescently labeled protein, the array is washed, and stable interactions are identified by scanning the slide for fluorescent spots (see Fig. 1). It is also possible to probe the array with an epitope-, hapten-, or biotin-tagged protein and to subsequently visualize bound proteins with a fluorescently labeled antibody or streptavidin conjugate. However, direct labeling of the protein of interest produces the best results. For a labeling and probing protocol for protein-protein interactions, see Protein Arrays: Labeling the Protein and Probing the Array for Protein-Protein Interactions.
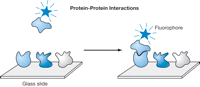
Protein-protein interactions. (Courtesy of Gavin MacBeath, Harvard University.)
Protein-Small Molecule Interactions
Protein arrays can also be used to screen for protein-small molecule interactions. For this application, a protein microarray is incubated with a fluorescently labeled compound, the array is washed, and stable interactions are identified by scanning the slide for fluorescence (see Fig. 2 and Protein Arrays: Labeling the Compounds and Probing the Array for Protein-Small Molecule Interactions). It is convenient to label compounds by covalently linking them to a carrier protein, such as bovine serum albumin (BSA), which has previously been labeled with a fluorophore. Not only is it often more convenient to label proteins in this way (rather than by directly coupling them to a fluorescent molecule), but the carrier protein also aids in rendering the labeled compound water-soluble. In addition, the valency of the conjugate can be increased to detect low-affinity interactions if desired. If the compounds cannot easily be immobilized, they may, in principle, be labeled with tritium and the interactions identified on the basis of radioactive spots. However, this approach has not yet been tested.
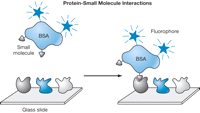
Protein-small molecule interactions. (Courtesy of Gavin MacBeath, Harvard University.)
Kinase-Substrate Interactions
In addition to screening for long-lived interactions, protein arrays can also be used to screen for transient interactions, such as those between an enzyme and its substrate. As long as the enzyme modifies its protein substrate and that modification can be detected, the transient interaction between enzyme and substrate can be inferred. Perhaps the most useful application in this area is the identification of putative substrates for protein kinases. Protein kinases modify their substrates by transferring a phosphate group from adenosine 5′-triphosphate (ATP) to a side chain on the protein (serine, threonine, tyrosine, or histidine). To identify substrates of protein kinases, a protein microarray is incubated with a kinase and [γ-33P]ATP (see Fig. 3). Following an appropriate incubation, the array is washed, coated with a photographic emulsion, further incubated, and finally developed and imaged with a scanning light microscope (for details, see Protein Arrays: Probing the Array and Detecting Radioactive Spots for Kinase-Substrate Interactions).
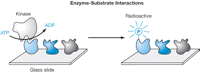
Enzyme-substrate interactions. (Courtesy of Gavin MacBeath, Harvard University.)
Protein Profiling
In addition to screening for interactions, protein arrays can be used to quantitate the abundance and modification states of proteins in complex mixtures (such as serum, cell culture supernatants, or even cell lysates). For this application, it is necessary to array receptor molecules, such as antibodies, on the slides and then use these molecules to specifically capture their cognate antigens from solution. Although various label-free detection methods are under development, it is likely that investigators will continue to rely on fluorescent labeling methods for some time. There are two ways to detect the captured proteins using fluorescent dyes. The direct method (see Fig. 4) relies on labeling all of the proteins in the complex solution with a fluorescent dye and then capturing the labeled proteins on the antibody array. The advantage of this method is that two different samples can be compared directly in a competitive binding experiment using two different colored dyes. The disadvantage of this approach, however, is that it is often difficult to label low-abundance proteins with high efficiency. Moreover, this approach requires extremely specific reagents to analyze low-abundance proteins (cross-reactivity of the reagents become a problem when the antigen is present at a much lower concentration than other proteins with similar epitopes).
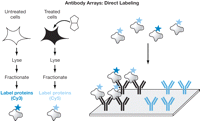
Antibody arrays: direct labeling. (Courtesy of Gavin MacBeath, Harvard University.)
The other way to detect the captured proteins does not require labeling the proteins themselves. Instead, each protein is captured by one reagent and then detected in a second step with a second reagent (see Fig. 5). The second reagent (antibody) recognizes the protein at a site that does not overlap with the recognition site of the first reagent (antibody). In this sandwich approach, the second reagent is labeled and thus provides the signal. The advantage of this approach is that it does not require labeling the proteins themselves. Moreover, additional specificity is gained by using two different reagents for each protein. The disadvantage, however, is that it is more difficult to assemble matched pairs of antibodies for each protein of interest. Although there are reasons to pursue both approaches, the sandwich approach is more appropriate for analyzing very complex solutions (cell lysates), and this method is detailed in Protein Arrays: Sandwich Approach for Analyzing Complex Solutions.
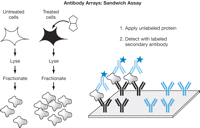
Antibody arrays: sandwich labeling. (Courtesy of Gavin MacBeath, Harvard University.)
Reading and Interpreting the Arrays
One of the advantages of constructing protein arrays on glass slides is that they can be scanned and analyzed using commercially available instrumentation. Several companies sell fluorescence slide scanners. In general, there are two types of scanners: those that use lasers to excite the fluorophores on the slide and a photomultiplier tube (PMT) to measure fluorescence and those that use a white-light source coupled with filters to illuminate the slide and a charge-coupled device (CCD) to measure fluorescence. Both types of instruments can be used to read protein arrays. The advantage of the PMT is that it is often faster and less expensive, whereas the advantage of CCD is that it tends to offer a higher signal-to-noise ratio and is more flexible with respect to the range of fluorophores that can be read.
All commercially available scanners come with image-processing software. At a minimum, the software enables the fluorescence of each spot to be quantified and the data to be exported to a spreadsheet. The analysis of protein array data may become more sophisticated in the future; however, at present, we have used relatively simple manipulations to interpret our results. For antibody array data, changes in fluorescence from one sample to the next are best expressed as fold changes. If purified antigens are available, standard curves can be generated, offering absolute, rather than relative, quantification.
Interaction data require more processing. One of the powerful aspects of protein arrays is that they can be used to determine the full n × n matrix of protein-protein interactions within a set of n proteins. To do this, n arrays of all n proteins are prepared, and each array is probed with a different member of the set. Each labeled protein, however, will produce a different level of background binding to the arrays and a different maximum signal for specific binding. As a result, it is necessary to apply some sort of normalization to determine which interactions are real and which are likely to arise from nonspecific binding. We have found it useful to quantitate the fluorescence of each spot on an array and then divide that intensity by the mean intensity of all the spots on the array to yield an “x-fold above the mean” value. An arbitrary decision must then be made as to how large an “x-fold above the mean” value is required to score the interaction as real.
As a further complication, some proteins on the array exhibit higher levels of nonspecific binding than others. If only a single array is processed, it is impossible to determine whether the signal at a given spot is due to a specific binding event or arises from nonspecific binding to an intrinsically “sticky” protein. One advantage of the n × n experiment is that it is possible to see how each immobilized protein interacts with all n-labeled proteins. Thus, a second level of confidence can be gained by analyzing each immobilized protein over the n different arrays. Each “x-fold above the mean” value for a given immobilized protein can be divided by the mean of the “x-fold above the mean” values on all n arrays. This effectively removes the nonspecific binding effect and highlights interactions that are more likely to be real.








