
Ultrastructural Immunochemistry
Adapted from “Ultrastructural Immunochemistry,” Chapter 7, in Immunohistochemistry: Methods Express (ed. Renshaw), from the Methods Express series. Scion Publishing Ltd., Oxfordshire, UK, 2006.INTRODUCTION
The use of colloidal gold technology was undoubtedly the most significant event in the development of immunochemistry. Gold particles are particularly useful for transmission electron microscopy (TEM) studies, because they scatter electrons strongly and even small particles are clearly visible under the electron microscope. Before proceeding to immunogold staining, it is important to gather as much information as possible about the antibody of interest and its respective antigen: Where is it likely to be located? Is the antigen extracellular, intracellular, membrane-associated, or a soluble component of the cytoplasm? Is it present in significant quantities? Is it sequestered at high concentration in any specific subcellular compartment, such as the mitochondria or the nucleus? How vulnerable to fixation and embedding is the antigen of interest? Information on the specificity of antibodies from Western blotting is valuable, but is not guaranteed to be useful for immunochemistry. Antibodies that “work well” on blots frequently have to be used at concentrations of up to three or more orders of magnitude greater for immunofluorescence and even more for immunogold staining studies, and some antibodies simply cannot be used for immunochemistry. This article describes methods and considerations for the use of immunogold staining, including fixation, controls, resolution, and quantification.
RELATED INFORMATION
The following protocols describe immunogold staining of various types of sections for TEM:
-
Immunogold Staining of Epoxy Resin Sections for Transmission Electron Microscopy (TEM) (Skepper and Powell 2008a)
-
Immunogold Staining of London Resin (LR) White Sections for Transmission Electron Microscopy (TEM) (Skepper and Powell 2008b)
-
Immunogold Staining Following Freeze Substitution and Low Temperature Embedding After Chemical Fixation or After Cryoimmobilization for Transmission Electron Microscopy (TEM) (Skepper and Powell 2008c)
-
Immunogold Staining of Ultrathin Thawed Cryosections for Transmission Electron Microscopy (TEM) (Skepper and Powell 2008d)
For the purpose of this article and the related protocols, the terms “immunochemical” and “immunogold” can be considered synonymous.
See also Detection of Gold-Labeled Reagents (Harlow and Lane 2006).
BACKGROUND
The landmark publication by Coons et al. (1941) demonstrated that an antibody conjugated to a fluorochrome retained its ability to recognize and bind tightly to its antigen. This was arguably the key event in the development of immunofluorescence microscopy. Once the general principles for immunochemical staining were established, the introduction of particulate markers for TEM followed quickly. Ferritin, a moderately electron-dense heme protein, was the first to be conjugated to antibodies some 18 years later (Singer 1959; Rifkind et al. 1964). In the pioneering study by Faulk and Taylor (1971), it was demonstrated that protein molecules, including antibodies, could be adsorbed onto the surface of gold particles with little or no loss of their biological activity. Next, the ability to produce colloidal gold particles with different mean sizes and non-overlapping size-frequency distributions (Frens 1973) brought the potential for immunochemical staining of multiple antigens on the same thin section, a development that significantly improved the value of the method. The application of random sampling strategies and the use of unbiased stereology allowed us to make quantitative comparisons of labeling density (Griffiths et al. 1993; Lucocq 1993). In some instances, it is possible to estimate the concentration of the antigen within its host tissue by making a comparison of labeling density on the specimen with that over an internal standard containing a known concentration of antigen (Storm-Mathisen and Ottersen 1990).
FIXATION AND ITS EFFECT ON ANTIGEN-ANTIBODY BINDING
The degree of resistance of the antigen to fixation is a key issue in immunochemical staining. In general, the stronger the fixative used, the better the ultrastructure of the thin sections. Unfortunately, the opposite generally applies to the ability of the antibody to bind its antigen. This also relates to cryotechniques. It is possible to freeze and embed tissue at low temperature without any chemical fixation, but ultrastructure is always compromised. If a chemical fixative is added to the substitution mixture, the frozen tissue is dehydrated and fixed at the same time. Fixation is less efficient at low temperature, but as a rule of thumb, the stronger the chemical fixation, the better the ultrastructural preservation and in particular that of membranes. The gold standard is to find the appropriate compromise that allows one to answer the biological question.
Glutaraldehyde and formaldehyde are the two fixatives in most common use, either individually or in combination. Formaldehyde is a monoaldehyde that interacts principally with proteins forming methylene bridges or polyoxymethylene bridges in a concentration-dependent manner. Glutaraldehyde is a dialdehyde that gives superior ultrastructural preservation but causes significant conformational changes to the tertiary structure of proteins. This frequently compromises the ability of an antibody to bind to its antigen. For a detailed discussion on the chemistry of fixation, see Griffiths et al. (1993), Hayat (1981), and Glauert and Lewis (1998). In this context, a “stronger” fixative will be regarded as a fixative containing higher concentrations of the reactive aldehydes.
The ability of the antibody to bind its antigen may be lost at several key stages of processing for TEM: during chemical fixation, dehydration in organic solvents, infiltration with epoxy or acrylic resin, or heat curing or polymerization of the resin. New antibodies should always be tested by a method that does not amplify signal, such as a species-specific, fluorescent secondary antibody method, before proceeding to electron microscopy. There are several key questions to ask if an antibody has been used for prior immunochemical studies:
-
1. Does the antibody work only on unfixed or cold acetone/alcohol-fixed cryostat sections or cell cultures? If the answer is yes, this antibody may only be usable in methods employing cryoimmobilization and freeze-substitution in pure organic solvent rather than after chemical fixation.
-
2. At what strength and duration of fixation will the antigens survive and still offer immunogenicity to bind to their respective antibodies?
-
3. Does the antibody work on sections of formalin-fixed, paraffin wax-embedded tissue, without antigen-retrieval treatment?
It is wise to undertake a systematic evaluation of fixation on a tissue known to contain significant amounts of the antigen under study. This constitutes a positive control, which is highly desirable, if not essential, in any rigorous study and may well provide critical information. Fixation of tissues and organs is best carried out by vascular perfusion (Hayat 1981; Glauert and Lewis 1998) to minimize the diffusion distance into the tissue for the fixative. There are, however, circumstances where fixation by perfusion is impossible or may be undesirable. Bendayan et al. (1987) showed that immunogold staining of serum albumin in glomerular capillaries was reduced dramatically after perfusion fixation, presumably because the serum albumin molecules were washed out during exsanguination.
If it is not possible to fix by perfusion, e.g., when working with human tissues from surgical material or biopsies, samples should be small. A simple method of achieving uniformity of fixation is to glue two safety razor blades together at the shank, to produce two parallel blades <1 mm apart. Tissues are sampled using a gentle slicing motion, rather than by applying significant vertical force, in order to minimize mechanical damage. Alternatively, a tissue chopper or vibrating microtome can be used to cut thin slices. Cells in culture are much easier to deal with, because diffusion distances for fixatives are minimal. They should be cooled to 4°C and rinsed in normal saline (0.9%, w/v, sodium chloride) before fixation. Nonadherent cells can be fixed in suspension, while adherent cells should be fixed in situ for 30-60 min and then scraped free of their substrate.
The temperature and duration of fixation should both be standardized to maintain uniformity between experiments. We carry out initial fixation tests at 4°C for no more than 120 min for tissues and 30-60 min for cell cultures, while others prefer fixation at 37°C (Peters et al. 2003), but only trial and error will determine the appropriate compromise between structural preservation and the ability of the fixed antigen to bind antibody. Safety is a major issue when fixation is carried out at temperatures above 4°C, because aldehydes are volatile, formaldehyde is a known carcinogen, and glutaraldehyde can cause occupational asthma. If fixation is performed at an elevated temperature, it should be carried out in a fume hood.
It is convenient to test new antibodies on adherent cell cultures expressing the antigen of interest, grown on glass coverslips, or on cryostat sections. Cells are grown to near-confluence on 19-mm diameter coverslips of No. 1 thickness and fixed for 30-60 min at 4°C. Naturally, if the antigens will survive longer periods of fixation (up to 4 h), then ultrastructural preservation will be even better. They are rinsed in four to six changes of buffer before being stained immunochemically. Alternatively, fixed tissues are infused with 20% (w/v) sucrose and frozen to prepare cryostat sections. We routinely store a range of fixed and unfixed tissues (myocardium, liver, gut, placenta, etc.) under liquid nitrogen so that material is always available for testing new antibodies. An initial test is carried out comparing the effects of weak and strong fixatives using the following solutions:
-
(a) 1% (w/v) formaldehyde in 0.1 M PIPES or HEPES buffer (pH 7.4) containing 3 mmol/L calcium chloride
-
(b) 4% (w/v) formaldehyde in 0.1 M PIPES or HEPES buffer (pH 7.4) containing 3 mmol/L calcium chloride
-
(c) 8% (w/v) formaldehyde in 0.1 M PIPES or HEPES buffer (pH 7.4) containing 3 mmol/L calcium chloride
-
(d) 3% (w/v) formaldehyde plus 0.05%-0.5% (w/v) glutaraldehyde in 0.1 M PIPES or HEPES buffer (pH 7.4) containing 3 mmol/L calcium chloride
The fixatives shown above are listed in ascending order of potential ultrastructural preservation but probable descending order of antibody binding. Tissue sections or cultured cells fixed in solutions (a), (b), and (c) are ready for immunochemical staining after rinsing in buffer. Those fixed in solution (d) must be incubated in 0.5% (w/v) sodium borohydride for 5-10 min and rinsed in buffer to quench the autofluorescence generated by glutaraldehyde. Test parameters should also include a range of dilutions of primary antibodies, usually 1:5, 1:25, 1:100, and 1:1000 for monoclonal antibodies and 1:50, 1:250, 1:1000, and 1:5000 for polyclonal antibodies.
Antigens that survive very strong fixation and embedding in paraffin wax may well survive ambient temperature dehydration and embedding in thermally cured or polymerized epoxy resin after secondary fixation with osmium tetroxide. In this method, the osmium tetroxide is removed from the superficial layers of the section by treatment with periodic acid and/or sodium metaperiodate (Skepper et al. 1998). Thin sections are floated on drops of the oxidizing agent of choice, e.g., 5% (w/v) sodium metaperiodate for 10-20 min, and then rinsed thoroughly with ultrapure H2O before commencing immunochemical staining. Periodic acid and sodium metaperiodate are both oxidizing agents with differing efficacies. Some workers use only sodium metaperiodate, while others suggest that a sequential treatment with both produces stronger immunochemical staining. We tend to use a single treatment with sodium metaperiodate, which removes osmium tetroxide from the surface of the thin section, and in some cases this will enhance the binding of an antibody to its antigen at that surface. Antigens that withstand modest fixation but not paraffin wax embedding are generally more suitable for embedding in acrylic resin at ambient or at low temperature. Antibodies that only work on unfixed cryostat sections may work in cells or tissues that have been cryoimmobilized, dehydrated by freeze-substitution, and embedded at low temperature. However, there is no guarantee that the integrity of the antigen will not be compromised by the subsequent dehydration, embedding, and curing or polymerization of the resin.
It may also be necessary to use a stronger fixative to retain antigens that are freely soluble in the cytoplasm (Crapo et al. 1992). It is interesting to note that cells with a high content of secretory granules and endoplasmic reticulum often show reasonable preservation, even after weak fixation, particularly if they are processed subsequently using the freeze-substitution and low-temperature embedding route. This may be at least partly due to their high protein content (see Fig. 1). As the strength of fixation is reduced, ultrastructural preservation becomes poorer, particularly that of membranes. The low-temperature methods compensate to some degree, but it is inevitable that weaker fixation means poorer preservation. The method that retains the best membrane preservation is undoubtedly the ultrathin, thawed cryosection or “Tokuyasu” method (Tokuyasu 1986), but again, stronger fixation gives better preservation. A comprehensive description of this technique is beyond the scope of this article, and the reader is referred to the text by Griffiths et al. (1993) and the seminal papers by Liou et al. (1996) and Peters et al. (2003).
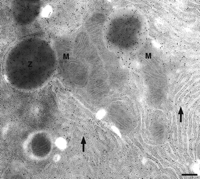
Thin section of a rat pancreatic acinar cell. The section was fixed in 3% formaldehyde, cryoprotected in 30% polypropylene glycol, dehydrated by freeze substitution, and low-temperature embedded in Lowicryl HM20. Cells were immunolabeled for the presence of amylase. Gold particles indicate the rough endoplasmic reticulum (arrows) and zymogen granules (Z). Mitochondria are unlabeled, showing that nonspecific labeling is low. Bar, 200 nm. (Reprinted with permission from Scion Publishing Ltd. © 2006.)
CONTROLS
Controls are essential but ostensibly simple, requiring tissue or cells expressing significant amounts of the antigen as a positive control. A negative control is equally important, as it will indicate whether there is nonspecific binding of primary or secondary antibodies. Sections of cells should also be exposed routinely to the secondary antibody alone to be certain that there is no nonspecific binding to any component of the tissue. In a recent unpublished study carried out with Raghu Padinjat of the Babraham Institute (Cambridge, UK), we encountered a most elegant example of a combined positive and negative control in adjacent cells of the same tissue. Ommatidia are the light-sensing structures of the Drosophila eye. Each ommatidium contains seven rhabdomeres (see Fig. 2), which have extensive membrane systems derived from microvilli. The membranes of six of the rhabdomeres are rich in rhodopsin, a light-absorbing pigment, while the seventh rhabdomere contains no rhodopsin (see Fig. 2), making it an ideal negative control. If excessive nonspecific binding of the primary or secondary antibody is apparent, protein can be added to the buffers to inhibit it competitively. Various proteins are used for this purpose, but in our hands bovine serum albumin (BSA) or coldwater-fish gelatin, both used at 0.5%-4% (w/v), give the most consistent results. Remember that this will also competitively inhibit specific binding, so the concentration of blocking protein should be kept as low as possible.
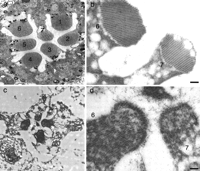
Thin sections through a single ommatidium from a wild-type or mutant Drosophila eye, immunolabeled for rhodopsin. The eyes were fixed in 3% glutaraldehyde, osmicated, and embedded in Spurr’s resin (a,b; wild-type) or fixed in 4% formaldehyde and embedded in LR White (c,d; mutant). (a) Each ommatidium contains seven rhabdomeres. (b) Rhabdomeres 1-6 express rhodopsin, while rhabdomere 7 does not. (c) In the mutant eye, ommatidia are deleted or altered. (d) The rhabdomeres are also structurally altered, but their staining pattern for rhodopsin remains unchanged, with no expression of rhodopsin in rhabdomere 7. Bars, 200 nm. (Reprinted with permission from Scion Publishing Ltd. © 2006.)
WHY DO WE NEED TO USE ELECTRON MICROSCOPY?
The answer to this is resolution. In the light, confocal, and two-photon microscopes, resolution is diffraction-limited to 180-200 nm in the x-y axes, dependent on the numerical aperture of the objective lens and the wavelength of light used to generate the image. Resolution in the z axis is much poorer at 500-600 nm. However, there are techniques that can bypass these limitations. These include total internal reflection fluorescence (TIRF) microscopy, stimulated emission depletion microscopy, and 4Pi microscopy. TIRF (Chung et al. 2006) and 4Pi (Egner et al. 2004) microscopy can exceed 100-nm resolution, but with severe limitations on specimen and lens geometry in 4Pi microscopy and in the depth of imaging into a sample with TIRF microscopy. Stimulated emission depletion microscopy (Willig et al. 2006) can exceed 50-nm resolution, but requires a very high signal-to-noise ratio and an almost ideal sample.
QUANTIFICATION
If a single compartment is being stained immunochemically and the biological question is simply whether or not there is staining over that compartment, then quantification is unnecessary. If label (staining) density is low and you wish to make a comparison between multiple compartments in control and experimental subjects, then quantification is essential. Quantification of label density is simple and strengthens data immensely. It is a simple extension of stereology, which is used to gain three-dimensional data from (effectively) two-dimensional sections. When comparing mutant and wild-type organs, it is desirable to start the comparison with an estimate of the volume or reference space of the organ itself. If the organ of the mutant is halved in volume but the percentage of it occupied by a specific cell type is doubled, the total volume of that cell type is unchanged. This phenomenon is known as the “reference trap” (Brændgaard and Gundersen 1986). A typical example might be to examine the effect of a mutation on the distribution of rhodopsin in the eye of Drosophila (see Fig. 2). After fixation and embedding in a suitable resin, serial sections (2 μm in thickness) are cut through the eye and the Cavalieri method (Howard and Reed 1998) is used to estimate the volume of the eye in mutant and wild-type flies. At four randomly selected levels through the layer containing the rhabdomeres, thin (50-70 nm in thickness) sections are cut, immunogold labeled for rhodopsin, and contrast counterstained with uranyl acetate and lead citrate. Both uranyl acetate and lead citrate impart contrast to the tissue. They are viewed at 80 kV in a transmission electron microscope using a 10- or 20-μm objective aperture to maximize contrast. A quadratic (square) lattice is overlaid on the TEM image and the volume fraction (Vv, expressed as a percentage) of the eye occupied by ommatidia and rhabdomeres is estimated by point counting (Howard and Reed 1998), i.e., counting the number of points (P) from the intersections of the counting lattice that overlay the area of interest (i). The formula for this calculation is:
Vvrhabdomere (%) = (Pirhabdomere/Pitotal) × 100
where Vvrhabdomere is the percentage of the eye occupied by rhabdomeres, Pirhabdomere is the number of lattice intersections overlaying rhabdomeres and Pitotal is the total number of lattice intersections overlaying all compartments of the eye. Therefore, if Pirhabdomere = 10 and Pitotal = 100, 10% of the eye is occupied by rhabdomeres.
The light-absorbing pigment rhodopsin is associated with the photoreceptor membranes of the rhabdomere, and immunogold label density can be calculated as the number of gold particles per unit area (number/square micrometer) of rhabdomere. Number/unit area can be estimated by randomly selecting squares of the counting lattice overlying the areas of interest (rhabdomeres) and counting all of the gold particles within the square and those within the frame that also intersect with two of the four boundary lines of the counting frame. This is known as the forbidden line rule (Gundersen 1977) and prevents the underestimation of particle density that occurs if particles are only counted if they are within the square but not touching the counting frame. Similarly, the number will be overestimated if all particles, including those intersecting all four boundaries, are included. The area of an individual square counting frame of a quadratic lattice is D2, where D is the distance between two intersections of the lattice. Therefore, gold label density can be estimated by summing the number of gold particles in, e.g., 10 randomly selected test frames and dividing that by the total area of those frames in micrometers.
The procedure described above gives a parametric estimate of gold labeling density over a structure or series of subcellular compartments. Mayhew and coworkers (Mayhew et al. 2002; Lucocq et al. 2004) have suggested a nonparametric alternative that estimates the “relative labeling density” between compartments and between control and experimental subjects.








