
Characterization of Protein–DNA Interactions Using Protein Microarrays
- 1Department of Pharmacology and Molecular Sciences, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
- 2The Center for High-Throughput Biology, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
- 3Department of Ophthalmology, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
- 4The Solomon H. Snyder Department of Neuroscience, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
- 5The Sidney Kimmel Comprehensive Cancer Center, Johns Hopkins University School of Medicine, Baltimore, MD 21205, USA
INTRODUCTION
Protein–DNA interactions (PDIs) are critical for many cellular processes. We present here a protocol for the identification of PDIs in vitro using protein microarray technology. The procedure involves double-stranding synthesized DNA oligonucleotides with a fluorescent-labeled primer, binding the labeled double-stranded DNA directly to the protein microarray, and analyzing binding of the resulting PDIs. This approach provides simultaneous identification of PDIs for thousands of proteins, and multiple carefully designed DNA probes can be tested in parallel, which enables a rapid mapping of PDIs on a proteome-wide scale.
MATERIALS
Reagents
Agarose gel (3%)
Annealing buffer for PDI (10×)
Bovine serum albumin fraction V, heat shock (BSA; Roche 03116999001)
DL-Dithiothreitol (DTT; Sigma) (1 M)
DNA ladders (20 bp) and 6× DNA loading dye (Fermentas SM1323)
dNTPs, high-purity solution (GE Healthcare 27-2035-02)
Illustra MicroSpin G-25 Columns (GE Healthcare 27-5325-01)
Oligonucleotides, high performance liquid chromatography (HPLC)-purified (Integrated DNA Technologies)
Phenylmethylsulfonyl fluoride (PMSF) (0.2 M)
Protease Inhibitor Cocktail Tablets, complete (Roche 11873580001)
SYBR Gold Stain (Invitrogen S11494)
Taq DNA polymerase and 10× Standard Taq Reaction Buffer (NEB M0273)
Wash buffer
Equipment
Aluminum foil
Centrifuge, benchtop, with microplate rotor (Thermo 75004375)
Coverslips, LifterSlip (Thermo Scientific 25X60I24789001LS)
Equipment for agarose gel electrophoresis
Filter, 0.45-µm (VWR)
Humidified chamber
-
Prepare the chamber by filling an empty pipette tip box with about half an inch of sterile water. Wipe the inside of the lid and the tip rack with 70% ethanol using laboratory tissues.
Image acquisition and analysis system
Laboratory tissues (e.g., Kimwipes)
Micro slide boxes (VWR 48444-004)
Microarray analysis software, GenePix Pro 6.0 (MDS Analytical Technologies)
Microarray scanner, GenePix 4000B (MDS Analytical Technologies)
Micropipettor and tips
Nalgene disposable sterilization filtration units, 0.2-µm filter (Fisher)
Pipette tip box, empty
Polymerase chain reaction (PCR) tubes
-
Alternatively, 96-well plates can be used in Step 5.
Protein microarrays (for details, see Discussion)
Shaker
Spectrophotometer, NanoDrop 2000 (Thermo Scientific)
Syringe, 1 mL (BD Biosciences)
Thermal cycler
Ultraviolet (UV) transilluminator
METHOD
Oligonucleotide Design and Synthesis (HPLC-purified)
-
These steps will take ~5 d to complete.
-
1. Synthesize oligonucleotides containing two to four tandem repeats (∼40 nt total) of a DNA motif plus a universal sequence at the 3′ end (5′-Motif-Motif-Motif-CCCTATAGTGAGTCGTATTAGGATCC-3′). This oligonucleotide is named Oligo-A.
Using multiple units per probe showed stronger binding signals than a single unit in our pilot assays.
-
2. Synthesize a Cy5-labeled universal oligonucleotide complementary to the universal sequence used in Oligo-A (5′-Cy5-GGATCCTAATACGACTCACTATAGGG-3′). This oligonucleotide is named Oligo-B.
-
3. Synthesize a pair of competitor oligonucleotides as Oligo-C (5′-AAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAACCCTATAGTGAGTCGTATTAGGATCC-3′), and Oligo-D (5′-GGATCCTAATACGACTCACTATAGGGTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-3′).
Double-Stranded DNA Probe Preparation
-
These steps will take ∼5 h to complete.
-
4. Dissolve all the synthesized oligonucleotides in H2O to a final concentration of 0.2 mM. Store at −20°C; make aliquots to avoid all-batch contamination and repeated freeze-thaw cycles.
Completely seal the tubes containing the Cy5-labeled DNA with aluminum foil and turn off the light during pipetting. Steps 4–6 must be performed so as to avoid light damage to the Cy5-labeled DNA.
-
5. Add the following reagents to a PCR tube:
-
7.5 µL of H2O
-
1 µL of 10× Standard Taq Reaction Buffer
-
0.4 µL of 10 mM dNTP
-
0.1 µL of Taq DNA polymerase
-
0.5 µL of 0.2 mM Oligo-A
-
0.5 µL of 0.2 mM Oligo-B.
-
Mix well by pipetting.
This step can be conducted in 96-well plates to increase the throughput of double-stranded DNA (dsDNA) production.
-
-
6. Place the PCR tube into a thermal cycler and run four cycles of the following two steps:
-
i. 55°C for 1 min
-
ii. 72°C for 10 min.
6. Place the tube on ice.
-
-
7. Meanwhile, generate double-stranded competitor DNA as follows:
-
i. Add 450 µL of 0.2 mM Oligo-C, 450 µL of 0.2 mM Oligo-D, and 100 µL of 10× annealing buffer for PDI to a PCR tube.
-
ii. Place the tube in a thermal cycler, heat at 95ºC for 5 min, and step down the temperature to 4ºC by 0.5ºC per 30 sec.
-
iii. Place the tube on ice.
-
-
8. Purify the dsDNA probe and competitor DNA by using Illustra MicroSpin G-25 Columns according to the manufacturer's protocol.
Avoid exposure of the Cy5-labeled DNA probe to light.
-
9. Perform DNA electrophoresis:
-
i. Dilute 1 µL of the purified dsDNA probe in H2O to 10 µL.
-
ii. Dilute 1 µL of the purified competitor DNA in H2O to 100 µL and aliquot 10 µL.
-
iii. Dilute 1 µL of 0.2 mM Oligo-B in H2O to 200 µL and aliquot 10 µL.
-
iv. Add 2 µL of 6× DNA loading dye to each of the diluted samples and mix well by pipetting.
-
v. Load the samples on a 3% agarose gel along with a 20-bp DNA ladder.
-
vi. Run the gel.
-
-
10. Stain the gel with SYBR Gold Stain according to the manufacturer's protocol. Visualize the DNA probe with a UV transilluminator and image acquisition and analysis system. Make sure that adequate dsDNA is generated and that it is of the expected length.
See Troubleshooting.
-
11. Quantify the concentrations of the dsDNA probe and competitor DNA by using a NanoDrop apparatus. Normalize the molar concentrations of the DNA probe and competitor DNA to 4 µM and 40 µM, respectively.
Avoid exposure of the Cy5-labeled DNA probe to light.
The purified dsDNA probe and competitor DNA can be kept overnight at 4°C or frozen for a week at −20°C.
Probing Protein Microarrays with DNA Probes
-
Blocking will take ∼3 h, followed by overnight incubation and 1 h of wash and scanning.
-
12. Prepare 200 µL of blocking buffer for each microarray. For 1 mL, combine:
-
500 µL of 2× base buffer
-
397 µL of H2O
-
3 µL of 1 M DTT
-
100 µL of 40 µM competitor DNA
-
Transfer the blocking buffer to a 1 mL syringe and filter through a 0.45-µm filter.
Prepare blocking buffer immediately before use and keep it on ice. Prepare one microarray for a negative control experiment with Oligo-B to identify nonspecific binding activities.
-
-
13. Overlay 200 µL of blocking buffer onto each microarray and cover the surface with a Lifterslip coverslip. Keep the microarray in a humidified chamber at 4°C for 3 h.
Up to four microarrays can be processed in parallel in a single humidified chamber. One may further increase the throughput after gaining experience with the protocol.
There are two raised bars at the edge of one side of the Lifterslip. Face the bar side of the Lifterslip to the microarray to provide separation and allow for even dispersal of solution between the microarray and coverslip. Lower the coverslip from one end of the microarray slowly to the other end with caution to avoid generating bubbles under the coverslip.
See Troubleshooting.
-
14. Prepare 200 µL of hybridization solution for each microarray with the following:
-
100 µL of 2× base buffer
-
72.4 µL H2O
-
4 µL of 50× protease inhibitor cocktail
-
0.6 µL of 1 M DTT
-
1 µL of 0.2 M PMSF
-
20 µL of 40 µM competitor DNA
-
2 µL of 4 µM Cy5-labeled dsDNA probe (or 2 µL of 4 µM Oligo-B for the negative control experiment)
-
Transfer the hybridization solution to a 1-mL syringe and filter through a 0.45-µm filter.
Prepare hybridization solution immediately before use and keep on ice. Completely seal the tubes containing the Cy5-labeled DNA with aluminum foil and turn off the light during pipetting. Steps 14–18 must be performed so as to avoid light.
-
-
15. Perform hybridization as follows:
-
i. Lift the barcode end of the microarray; the Lifterslip will easily slide off along the length of the microarray at the short end.
-
ii. Drain the blocking buffer by tapping the slide sideways on laboratory tissues for 5–10 sec.
-
iii. Immediately overlay 200 µL of hybridization solution onto the microarray and cover with a new piece of Lifterslip.
-
iv. Keep the microarray in a humidified chamber at 4°C overnight (seal the chamber with aluminum foil).
DO NOT let the slide dry by leaving it out of buffer for too long, as dryness will cause denaturing of the proteins and hence, loss of binding signals. Face the bar side of the Lifterslip to the microarray. Avoid generating bubbles under the coverslip.
See Troubleshooting.
-
-
16. Lift the barcode end of the microarray and the Lifterslip will slide off along the length of the microarray. Immediately wash the microarray with 50 mL of prechilled wash buffer in an empty pipette tip box at 4°C for 5 min with gentle shaking.
DO NOT let the slide dry by leaving it out of buffer for too long. Seal the wash box with aluminum foil to avoid light.
-
17. Put the microarray vertically into a micro slide box with laboratory tissues at the bottom. Centrifuge the box in a benchtop centrifuge for 3 min at 1000g.
Do not centrifuge the slides at higher speed, as it might cause damage to the glass.
-
18. Scan the microarray with a GenePix 4000B scanner with 5-µm resolution detection at 635 nm. Ensure that no spots exhibit saturated signal intensities (median pixel intensity = 65,536). Save the scanned images as TIF files.
All slides that will be compared should be scanned using the same photomultiplier tube (PMT).
See Troubleshooting.
Data Quantification and Analysis
-
The duration of these steps is highly variable, and is dependent on the density of the arrays.
-
19. Align the GenePix Array List file (.gal) containing spot coordinates and protein names to the array image and obtain the foreground and background intensities of all the protein spots on the microarray using GenePix Pro 6.0. Save the intensity file as a GenePix Result file (.gpr) or export it as an Excel file.
The alignment can be achieved automatically by using the “program hot keys” in GenePix Pro 6.0. For example, Key F8 is used to “Find Array, Find All Blocks, Align Features”, and Shift + F5 to “Align Features in All Blocks”. However, the automatic alignment is not always accurate and it is necessary to manually inspect all the blocks and features to correct the inaccurate alignment for higher quality data acquisition. In the “Block Mode” (hot Key B), the blocks can be selected and moved by using mouse or arrow keys to roughly match the protein spots on a microarray image according to the position of positive control proteins. Then, in the “Feature Mode” (hot Key F), the position and size of each feature circle can be moved and adjusted to exactly match the corresponding protein spot. For spots with irregular shapes, select the block(s) containing the irregular spots, open “Options” (Alt + I), click the “Alignment” tab, check “Find irregular features” to set up parameters, and, finally, click “OK” and press F5. The irregular features should be aligned automatically. Change the parameters until the alignment is satisfactory.
-
20. Quantify the signal intensity of each protein spot by dividing the median foreground intensity by its median background intensity.
-
21. Normalize signal intensities of a set of spots within a block on a microarray by setting the median intensity of that block equal to one. A sliding window approach (such as ProCAT software employs) is preferable for within chip normalization (Zhu et al. 2006b).
Normalization is important to reduce spatial artifacts on the microarrays that might arise from uneven mixing of probes during binding, from uneven washing, or from uneven drying of the slide. Given the fact that proteins are randomly printed and only a small portion of proteins on a microarray bind to a target DNA sequence, it is reasonable to assume that the median signal intensity of each block on the microarray is one. Therefore, signal intensity of a spot (I) can be normalized as follows:
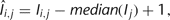 where î is the signal intensity after normalization, i is the protein index in a block, and j is the block index.
where î is the signal intensity after normalization, i is the protein index in a block, and j is the block index. -
22. Rank the normalized signal intensity of all the proteins and determine a cutoff value to identify positive hits, which are proteins binding to the DNA probe.
In some cases, spots can show strong binding signals that make the standard deviation (SD) value of the residing block very large and may reduce the sensitivity of detecting positive hits in the same block. Therefore, removal of such outliers is necessary before determining the SD values. We have previously proposed a method for identifying outliers. We compared histogram distributions of signal intensities above and below the mean value and then identified spots with extremely high signal intensity (Hu et al. 2009).
-
23. Identify nonspecific binding proteins that bind to Oligo-B from the negative control experiment and exclude them from the previous positive hits.
-
24. If multiple binding experiments are performed, use several DNA motif discovery tools to compare the results for identifying the optimal consensus sites (D'Haeseleer 2006).
TROUBLESHOOTING
Problem: There is a slight Oligo-B band in the DNA probe lane, which will generate false positive signals on the microarray.
[Step 10]
Solution: The concentration of Oligo-A may be lower than that of Oligo-B. Discard the probe. Increase the volume of Oligo-A in Step 5 and repeat Steps 5, 6, and 8–10.
Problem: Bubbles appear under the coverslip.
[Step 13,15]
Solution:The coverslip is placed improperly. Consider the following:
-
1. Gently tap the humidified chamber against a hard surface until bubbles move out of the protein-printed area.
-
2. If bubbles are hard to remove, lift the slip and readjust.
Problem: Signals are faint on the entire microarray.
[Step 18]
Solution: The DNA motif could be binding to proteins on the array with low affinity or with a fast dissociation rate. Repeat the binding assay with a higher DNA concentration and/or shorter wash time.
Problem: Strong signals are observed for too many proteins.
[Step 18]
Solution: The DNA motif could be binding to proteins on the array nonspecifically with high affinity. Repeat the binding assay with a lower DNA concentration and/or longer wash time.
Problem: Speckles and dust particles are visible in the scan.
[Step 18]
Solution: Consider the following:
-
1. Containers or vessels used to store and prepare wash buffer may not be clean. Make sure that these containers are cleaned thoroughly.
-
2. Filter wash solutions before use.
DISCUSSION
PDIs mediate a broad range of cellular processes, such as transcription, DNA recombination, DNA replication, DNA damage repair, and chromatin remodeling. A systematic analysis of PDIs in model organisms should contribute essential information toward mapping the genetic regulatory networks that regulate cell function. Traditionally, intensive effort has been invested in identifying DNA binding sites for transcription factors (TFs), the most important functional category of DNA-binding proteins (Vaquerizas et al. 2009). However, far less is known about the DNA binding specificity of other functional categories of DNA binding proteins that are not TFs.
Recent years have witnessed a rapid development of various high-throughput methodologies to profile TF binding sequences. The systematic evolution of ligands by exponential enrichment and serial analysis of gene expression (SELEX-SAGE) method enables in vitro selection of the optimal binding site of a TF (Roulet et al. 2002). However, this approach may be difficult to apply for analysis of large numbers of DNA binding proteins. Chromatin immunoprecipitation (ChIP), in which chromatin bound to a TF is immunoprecipitated, has been a standard method to characterize in vivo PDIs (Kuo and Allis 1999). When coupled with comprehensive deconvolution of the DNA sequences ChIPed with a TF, either by hybridization to a DNA microarray (ChIP-chip) (Ren et al. 2000) or via deep sequencing (ChIP-seq) (Robertson et al. 2007), it offers an unbiased, genome-wide methodology for identification of in vivo PDIs. Although powerful, it is technically challenging and highly dependent on the availability and quality of antibodies against a TF in question. This is particularly true in higher eukaryotes in which, unlike in yeast, genome-wide homologous recombination to endogenously tag every open reading frame (ORF) is not feasible. Moreover, it is often difficult to precisely pinpoint the consensus sequence for a TF only using the ChIPed DNA sequences.
To be able to precisely map a consensus sequence of high quality, Bulyk and colleagues have developed protein-binding DNA microarray (PBM) technology, in which a purified DNA-binding protein is directly probed to a double-stranded DNA microarray that contains all possible 10-base pair (bp) DNA sequences (Berger et al. 2008). The binding signals can then be detected and quantified by using fluorescently labeled antibodies. Because this method allows an exhaustive search of the entire 10-bp DNA space, a probe protein can usually recognize a cluster of DNA sequences of high homology among which subtle changes in binding affinity due to sequence variations can be easily detected and, therefore, a high-quality consensus site can be readily deduced. Similarly, the yeast or bacterial one-hybrid approach allows a systematic survey of 10- to 15-mer DNA space via various reporter systems (Deplancke et al. 2004; Meng et al. 2005). Like the SELEX approach, huge pools of clones have to be screened and the deconvolution of positive clones is dependent on large-scale DNA sequencing. The PBM and one-hybrid technologies have been applied to determine consensus sequences of TFs in mice, worms, and flies (Deplancke et al. 2006; Berger et al. 2008; Noyes et al. 2008). Additional technological developments include a microwell-based system that can be used to directly measure the affinity of a TF, or any DNA-binding protein, to any DNA sequence (Hallikas et al. 2006). However, this method requires prior knowledge of at least one high-affinity DNA-binding site for the protein of interest.
Alternatively, the use of protein microarrays is a particularly powerful method to identify proteins that can specifically recognize a DNA motif of interest, as a large number of individually purified proteins can be queried simultaneously in a microarray format (Hu et al. 2009). This approach can be viewed as complementary to the use of PBM or one-hybrid technologies. Recent advance in developing genome-wide ORF collections has led to the fabrication of functional protein microarrays for complex eukaryotic proteomes (Zhu et al. 2001; Hu et al. 2009). Functional protein microarrays, also known as protein chips, are constructed by immobilizing individually purified or via capture of in situ expressed proteins on a glass slide at high-density (Zhu et al. 2001; Tao et al. 2007). A wide range of assays have been developed for protein microarrays to probe various types of protein biochemical properties that enable unbiased, high-throughput screenings of thousands of proteins in a single experiment. Protein microarrays are an outstandingly flexible tool for high-throughput studies. Well established applications of protein microarrays include binding assays that detect protein–protein (Zhu et al. 2001; Jones et al. 2006), protein–small molecule (Huang et al. 2004), protein–DNA (Hall et al. 2004; Ho et al. 2006; Hu et al. 2009), protein–RNA (Zhu et al. 2007), antigen–antibody (Zhu et al. 2006a; Hu et al. 2007; Chen et al. 2009; Song et al. 2010), lectin–glycan (Kung et al. 2009), and lectin–cell interactions (Mahal 2008; Tao et al. 2008). Moreover, functional protein microarrays have also been applied to identify substrates of various types of protein posttranslational modifications, such as phosphorylation (Zhu et al. 2000; Ptacek et al. 2005; Schnack et al. 2008), ubiquitylation (Lu et al. 2008), acetylation (Lin et al. 2009), SUMOylation (Oh et al. 2007), and nitrosylation (Foster et al. 2009).
In a recent study, we have accomplished a large-scale analysis of human PDIs using protein microarrays (Hu et al. 2009). The goals were both to determine which proteins (e.g., TFs) could specifically recognize predicted DNA motifs and to discover unconventional DNA-binding proteins (uDBPs) in the human proteome. After searching the literature and available public databases, we collected 400 predicted and 60 known DNA motifs to perform the DNA-binding assays. The protein chip used for these studies included not only ∼90% of the annotated TFs, but also a wide range of other protein families, such as transcription co-regulators, nucleotide-binding proteins, RNA-binding proteins, chromatin-associated proteins, mitochondrial proteins, and protein kinases. A total of 4191 unique human proteins in full-length were expressed and purified from yeast to fabricate the protein microarrays and they were subsequently probed with the 460 fluorescently labeled DNA motifs. A total of 17,718 PDIs were identified. Among them we recovered many known PDIs and a large number of new PDIs for both well characterized and predicted TFs. Actually, >40% of the TFs showed specific DNA-binding activity. Using algorithms developed by our own group, we were able to determine new consensus sites for >200 TFs, which doubled the number of the previously known consensus sites (Hu et al. 2009; Xie et al. 2010).
Surprisingly, we observed that over 300 uDBPs, which were previously not known to specifically interact with DNA molecules, showed sequence-specific PDIs. These uDBPs include RNA-binding proteins, chromatin-associated proteins, nucleotide-binding proteins, mitochondrial proteins, and protein kinases, which raised an intriguing hypothesis that many human proteins may specifically recognize particular DNA sequences as a moonlighting function. To further explore whether the DNA-binding activities observed for these uDBPs were physiologically relevant or in vitro artifacts, we set out to determine the potential molecular mechanism behind the DNA-binding activity of a well-studied MAP kinase, Erk2. Using a series of in vitro and in vivo assays, such as gel shift, luciferase reporter assays, mutagenesis, and ChIP, we demonstrated that Erk2's DNA-binding activity is independent of its protein kinase activity and it acts as a transcription repressor in the interferon gamma signaling pathway (Hu et al. 2009). The study illustrates the power of protein microarrays for unbiased identification of PDIs in a proteome-wide scale and suggests that a moonlighting function of sequence-specific DNA-binding activity by uDBPs is probably a widespread phenomenon in humans.
ACKNOWLEDGMENTS
We thank the National Institutes of Health (NIH) (R01 GM076102) for funding support.
Footnotes
- © 2011 Cold Spring Harbor Laboratory Press








