
Imaging Synaptic Vesicle Recycling by Staining and Destaining Vesicles with FM Dyes
Adapted from Imaging in Neuroscience (ed. Helmchen and Konnerth). CSHL Press, Cold Spring Harbor, NY, USA, 2011.Abstract
The synaptic vesicle is the essential organelle of the synapse. Many approaches for studying synaptic vesicle recycling have been devised, one of which, the styryl (FM) dye, is well suited for this purpose. FM dyes reversibly stain, but do not permeate, membranes; hence they can specifically label membrane-bound organelles. Their quantum yield is drastically higher when bound to membranes than when in aqueous solution. This protocol describes the imaging of synaptic vesicle recycling by staining and destaining vesicles with FM dyes. Nerve terminals are stimulated (electrically or by depolarization with high K+) in the presence of dye, their vesicles are then allowed to recycle, and finally dye is washed from the chamber. In neuromuscular junction (NMJ) preparations, movements of the muscle must be inhibited if imaging during stimulation is desired (e.g., by application of curare, a potent acetylcholine receptor inhibitor). The main characteristics of FM dyes are also reviewed here, as are recent FM dye monitoring techniques that have been used to investigate the kinetics of synaptic vesicle fusion.
MATERIALS
Reagents
FM dye(s)
-
Dye concentrations are usually 2–10 µm for FM1-43 or FM4-64, and 25–40 µm for FM2-10. The dyes readily dissolve in water; stocks of 1–5 mm concentration should be prepared and can be stored at 4°C.
Neuronal tissue or cell sample of interest
Equipment
Imaging setup
-
An upright epifluorescence microscope equipped with a 40×–60× water-immersion objective is optimal. When working with cell cultures, the advantages of high-resolution oil-immersion objectives (e.g., 100×/1.4 numerical aperture [NA]) can be exploited by using an inverted setup. For FM1-43, maximal fluorescence is at 465-nm excitation/560-nm emission (Henkel et al. 1996); a conventional fluorescein filter set (435-nm excitation) can also be used. Because the major concern in FM imaging is phototoxicity (which appears usually before any clear signs of photobleaching and alters the ability of the preparations to recycle vesicles), keep the illumination to a minimum by including neutral-density filters. For example, when using a 40×/0.75-NA water-immersion objective with a 100-W mercury lamp, 5%–15% transmission is sufficient. On confocal setups, an argon 488-nm laser line is typically used for excitation, but special care has to be taken to avoid phototoxicity. Images are typically acquired by charge-coupled device (CCD) cameras or photomultiplier tubes, which allows for fast acquisition rates and software-based quantitative image analysis.
Neuronal stimulator (see Step 3)
METHOD
-
The general procedure for staining synaptic vesicles presented is outlined in Figure 1B. Dissection and experiments should be performed under the optimal physiological conditions for the experimental preparation. If stimulation is performed by high-K+ depolarization, the bathing solution should be adjusted by replacing KCl for NaCl to reach the desired final K+ concentration (typically 60 mm).
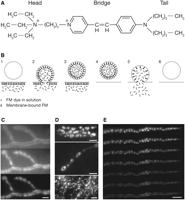
(A) Chemical structure of FM1-43. The molecule is composed of a positively charged head group, which prevents the dye from permeating membranes, and a lipophilic tail, which ensures dye partitioning into membranes. These components are connected by a double bond bridge, the number of double bonds determining the spectral characteristics. (B) FM dye staining and destaining. (1) The dye is added to the solution bathing the preparation. It does not have access to the intracellular space. (2) The preparation is stimulated. Vesicles exocytose and come in contact with the dye. (3) Vesicles endocytose and take up dye. (4) The dye is washed from the extracellular solution; it departitions from the plasma membrane but remains trapped in internalized vesicles (staining). (5) The stained preparation is stimulated in the absence of the dye. Vesicles fuse with the membrane and release dye (destaining). (6) Destained vesicles are internalized; a new experiment of staining and destaining can be performed. (C) Frog motor nerve terminal shown during staining. Images were acquired and processed identically, and are, therefore, directly comparable. (Top) The preparation had been bathed in FM dye-containing solution for 1 min. The surface membrane of the nerve terminal is faintly stained. (Middle) After a 1-min tetanus (30 Hz), followed by 15 min of rest in dye-containing solution, the terminal is brightly stained, but background fluorescence is also high. (Bottom) After dye washout, the background staining is greatly reduced, and clusters of vesicles can be observed as distinct fluorescent spots. (D) FM-labeled nerve terminals in different preparations: (Top) Drosophila larval NMJ. Scale bar, 3 µm. (Middle) Lizard (Anolis) NMJ. Scale bar, 2 µm. (Bottom) Cultured hippocampal neurons (fluorescent spots represent single presynaptic boutons). Scale bar, 5 µm. (Photograph courtesy of Dr. V. Murthy.) (E) Nerve terminal destaining. A stained preparation was stimulated at 2 Hz for ∼40 min in the presence of 10-µM curare to prevent muscle movement. The images were taken approximately every 5 min. The nerve terminal loses most of its initial fluorescence. Scale bar, 5 µm. (E, Reprinted, with permission, from Betz and Henkel 1994, © The Rockefeller University Press.)
Sample Preparation
-
1. Prepare neuronal tissue or cell sample. For NMJs, clean the muscle preparation by removing surface connective tissue, being careful not to damage muscle fibers or nerve cells (FM dyes intensely stain intracellular membranes of injured cells, creating unwanted background fluorescence). For cultured hippocampal neurons, cultures with minimal astrocytic background are preferable.
Staining
-
2. Wash the dye-containing solution onto the preparation. Incubate for ∼1 min, to allow the dye to diffuse into the synaptic clefts.
-
The dye stains the surface membranes of muscle cells (in NMJ) and nerve terminals (typical image shown in top panel of Fig. 1C, frog NMJ).
-
-
3. Stimulate the preparation in the presence of the dye. This will cause the preparation to fill with dye as vesicles are retrieved after ongoing exocytosis.
-
The increased fluorescence intensity is proportional to the number of exocytosed vesicles. Electrical tetanic stimulation of the nerve (30 Hz for 1–5 min) triggers recycling of 30%–80% of all synaptic vesicles in frog NMJ, ensuring adequate staining. High-K+ (60-mM) treatment of the preparation results in similar levels of dye uptake.
-
-
4. Allow the preparation to recover while still in the presence of the dye, to ensure complete recycling of the vesicles.
-
The length of the recovery period depends on the length of the stimulus—for example, 1–3 min tetani require 10–20 min of rest. Figure 1C (middle panel) shows the same nerve terminal after stimulation for 1 min (30 Hz), followed by 15 min of recovery. The terminal took up dye, and it appears much brighter than in the top panel. However, as the dye is still present in the chamber, background fluorescence is also high.
-
-
5. Wash the dye from all surface membranes, for 30–60 min, at 4°C.
-
Washing at low temperatures prevents dye release by spontaneous exocytosis. Dye-scavenging compounds, such as ADVASEP-7 (see Discussion), can be used to shorten washing times.
-
-
6. Image the preparation.
-
As can be seen in Figure 1C (bottom panel), fluorescent spots are now visible within the nerve terminal, and background fluorescence is low. The spots (usually 1–3 µm in diameter) represent clusters of synaptic vesicles containing a few hundred to several thousand vesicles. Most synapses investigated to date stain with FM dyes in a similar manner (see several examples in Fig. 1D), although the synaptic morphology may vary substantially. Note, for example, the ring-like appearance of the Drosophila larval NMJ boutons, which is caused by their vesicles being concentrated at the bouton periphery.
-
Destaining
-
7. Stimulate labeled preparations in the absence of the dye. On synaptic vesicle exocytosis, the dye will be released and will diffuse into the aqueous solution.
-
Figure 1E shows images from a time-lapse sequence from an experiment in which a frog NMJ nerve terminal was stimulated for 40 min at 2 Hz, losing most of its FM dye. Tetanic stimulation causes faster dye release than low-frequency stimulation (stimulation at 30 Hz for 2–3 min releases similar amounts of dye as 2-Hz stimulation for tens of minutes). During destaining, minimize the intensity of the excitation light to avoid phototoxicity without compromising image quality. A good rule of thumb is to adjust both the intensity with neutral-density filters and the exposure time such that the dynamic range of the initial (predestaining) images is ∼8 bits (meaning that the brightest pixel is ∼250–300 counts brighter than the dimmest one). Additionally, the number of images acquired should be kept as low as possible.
-
DISCUSSION
Release of neurotransmitters from nerve terminals requires the fusion of synaptic vesicles with the presynaptic plasma membrane (exocytosis). Next, the vesicles are internalized (endocytosis) and directed to the pool of vesicles inside the nerve terminal (vesicle recycling). As a consequence of this vesicle cycle, the inner leaflet of the vesicle membrane is temporarily exposed to the outside space, which provides an opportunity for membrane-staining compounds to specifically label recycling vesicles. Because the endocytic process also results in the uptake of a volume of fluid, even fluid-phase tracers can be used. Indeed, various markers have been used, such as sulforhodamine (Lichtman et al. 1985), fluorescently labeled antibodies (Kraszewski et al. 1996; Westphal et al. 2008), styryl dyes (i.e., FM dyes; Betz and Bewick 1992), and fluorescent dextrans (Holt et al. 2003). Among these markers, FM dyes have proved to be valuable in a number of different preparations, including neuromuscular junctions (NMJ) from the frog (Betz and Bewick 1992), snake (Teng et al. 1999), lizard (Lindgren et al. 1997), and larval Drosophila (Ramaswami et al. 1994); hippocampal neurons in culture (Ryan et al. 1993) and acute slice (Zakharenko et al. 2001); goldfish retinal bipolar cells (Zenisek et al. 2000); Caenorhabditis elegans neurons (Kay et al. 1999); rat calyx of Held (de Lange et al. 2003); inner and outer hair cells (Griesinger et al. 2002; Kaneko et al. 2006); and a variety of nonneuronal preparations (for a review, see Cochilla et al. 1999).
FM Dyes: Properties and Mechanism of Action
Several FM dyes have been synthesized (Betz et al. 1996), the most widely used being FM1-43 (chemical structure shown in Fig. 1A). Because of its amphipathic nature, the FM dye reversibly partitions into lipid membranes without permeating them, so that when dye is present in the bathing solution, some of it becomes trapped in endocytic organelles. Because the quantum yield of FM dye is several 100-fold higher when in membranes versus aqueous solution, these dyes are exceptionally well suited for labeling membrane-bound compartments.
All FM dyes share the same general structure: a positively charged hydrophilic head group and a lipophilic tail (usually formed by two aliphatic hydrocarbon chains) connected by a double bond bridge (Fig. 1A). The length of the tail determines the dye’s hydrophobicity and its ability to be washed out of membranes (e.g., the time constants for dye washout are ∼1sec for the short-tailed FM2-10, and 3–6 sec for FM1-43; see also Fig. 2A). The length of the double bond bridge connecting the two aromatic rings (i.e., the extent of the conjugated double bond system) determines the dye’s spectral properties. Dyes with one double bond (FM1-43, FM2-10) fluoresce at shorter wavelengths (green) than ones with two or three double bonds (FM4-64, red).
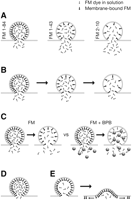
Approaches to imaging exocytosis with FM dyes. (A) FM dye release kinetics depend on dye hydrophobicity. FM dyes with lower hydrophobicity (FM1-84 > FM1-43 > FM2-10) departition faster from the membrane on exocytosis. (B) Partial FM dye release in kiss-and-run. Exocytosis through a transient kiss-and-run fusion pore releases only a fraction of the dye, leaving partially filled vesicles to release the remaining dye in successive rounds of fusion. (C) Probing partial FM dye release by quenching. If partial dye release occurs as a consequence of kiss-and-run, vesicles retain some dye after exocytosis (left). If the hydrophilic quencher bromophenol blue (BPB) is included in the bathing solution (right), it quenches any dye remaining in the vesicles (including any membrane-bound dye molecules), leading to a difference in fluorescence (when compared with the situation without BPB). For full collapse with complete loss of dye, application of BPB would not lead to a difference in fluorescence. (D) Complete dye retention. As shown by experiments combining electrophysiology and imaging, a fraction of vesicles might release a neurotransmitter, without the ability to lose or to take up FM dye. These results were interpreted in favor of exocytosis through a restrictive fusion pore. (E) Direct imaging of exocytosing vesicles by total internal reflection microscopy revealed little FM dye release into the fluid, but rather a lateral diffusion in the plasma membrane (with the dye later departitioning from the membrane at a distance from the point of exocytosis), indicative of full collapse fusion.
A typical FM dye experiment at a synapse is illustrated in Figure 1B. Preparations are bathed in a solution containing the dye, which labels the extracellular membranes (Fig. 1B1). Exocytosis is then induced (e.g., by electrical stimulation), and inner leaflets of vesicles come in contact with the dye-containing solution, thus becoming labeled (Fig. 1B2). After a fraction of the vesicles are internalized through compensatory endocytosis (Fig. 1B3), the dye is washed from the solution (Fig. 1B4), leaving label only within those organelles that formed while the dye was present. Exocytosis can then be monitored by stimulating these preparations in the absence of dye. The fluorescent molecules wash out of the vesicles into the bath, which also dramatically lowers quantum yield (Fig. 1B5) so that the decrease in fluorescence gives a direct measurement of vesicle release. After internalization of destained vesicles (Fig. 1B6), the preparation is ready for another round of staining and destaining.
A protocol is also available for FM Dye Photoconversion for Visualizing Synaptic Vesicles by Electron Microscopy (Hoopman et al. 2012).
Slice Preparations
FM studies in preparations such as acute brain slices are difficult because damaged membranes soak up a large amount of FM dye, which is then hard to wash out. This problem can be alleviated by quenching the fluorescence of the FM dye that is located outside of the membrane-bound organelles. Pyle et al. (1999) applied sulforhodamine to FM1-43-labeled hippocampal slices and obtained a rapid decrease in background fluorescence. This technique requires that the sulforhodamine be present throughout the imaging experiments because the FM dye is not washed from the slice. Another approach to improve the image quality has been to increase the amount of dye that is washed out of the preparation by including the oligosaccharide ADVASEP-7 during the washing steps. ADVASEP-7, which is a derivative of β-cyclodextrin, has a higher affinity for FM dyes than do biological membranes (Kay et al. 1999). The addition of ADVASEP-7 to washing solutions greatly reduces background staining, making it possible to image systems such as rat brain and C. elegans slices. Zakharenko et al. (2001) used this technique in acute hippocampal slices and showed an increase in presynaptic activity (increased rates of FM destaining) during long-term potentiation (the first such description of this phenomenon in a noncultured preparation).
Using FM Dyes to Image the Fine Dynamics of Exocytosis
Presynaptic boutons of hippocampal neurons in culture are the most frequently used preparation for studying vesicle recycling in central nervous system synapses. At the frog NMJ (see Fig. 1C,E), most vesicles recycle through a slow endocytic pathway (Richards et al. 2000), which relies on endosome-like intermediates and clathrin-dependent endocytic vesicle formation (Heuser and Reese 1973). Such a slow pathway may represent a significant bottleneck for vesicle recycling under conditions of high activity, especially for the hippocampal synapses, which typically contain just 100–200 vesicles. Thus, an alternative mechanism has been proposed, known as the kiss-and-run pathway (Fesce et al. 1994), in which vesicles transiently fuse with the plasma membrane through a relatively small pore, and endocytosis occurs simply by closure of the pore and retraction of the vesicle. A kiss-and-run mechanism might be able to recycle vesicles much more quickly than could the classical pathway. However, in hippocampal synapses, there was little evidence supporting fast recycling until investigators began using FM dyes. Some examples follow.
In full fusion, the vesicle membrane completely collapses into the plasma membrane so that FM dye departitions into the bathing solution, leaving behind a dye-free membrane to be endocytosed. As kiss-and-run is thought to only involve a transient fusion of the membranes, FM dye release on fusion should only be partial. A number of experimental schemes has been devised to exploit this concept.
One experiment focused on the release of different FM dyes from vesicles during destaining (Klingauf et al. 1998; Pyle et al. 2000; Fig. 2A). The destaining behavior of dyes with different hydrophobicities (the hydrophilic FM2-10 and the lipophilic FM1-43) was compared. Vesicles collapsing into the plasma membrane are expected to release both of the dyes completely and with similar kinetics. However, if the process of endocytosis is faster than the departitioning of the dye from the membrane, proportionally more FM2-10 should be released, which was indeed the observed result. Furthermore, the more lipophilic FM1-84 was released even more slowly than FM1-43 (Klingauf et al. 1998), suggesting that hippocampal boutons rely on fast vesicle recovery. One should note, however, that this differential release of dyes is not necessarily a general phenotype for all preparations (see the frog NMJ, Richards et al. 2000).
Aravanis et al. (2003) labeled a single vesicle per hippocampal bouton with FM1-43. Stimulating at low frequency, they mainly observed a partial loss of fluorescence per fusion event, indicating that the vesicle retained much of the dye after fusion. They also observed that individual vesicles fused repeatedly during the experiment (Fig. 2B). Complete loss of fluorescence intensity, indicative of full collapse fusion, was only seen rarely after a single action potential (but compare Chen et al. 2008, in which only evidence for full collapse was obtained).
Another elegant way of testing kiss-and-run fusion by use of FM dyes was devised by Harata et al. (2006). They labeled hippocampal cultures with FM1-43 and destained in either the presence or the absence of BPB (a small hydrophilic quencher; Fig. 2C). In this experiment, one expects to find different destaining behaviors depending on the fusion mechanism at work: For full collapse, all the FM dye will escape the vesicle and, hence, the presence of BPB will not affect the fluorescence signal. For kiss-and-run, some amount of dye is retained in the vesicle so that the presence of BPB causes a decrease in the fluorescence signal. The results suggested that at least a fraction of the vesicles retain a fraction of their FM dye contents during exocytosis.
Stevens and Williams (2000) combined imaging of FM2-10 and FM1-43 with electrophysiological recordings, stimulating vesicle release by hyperosmolarity. During hyperosmolarity-induced release, a proportion of the vesicles could have released glutamate, but neither lost nor took up dye (Fig. 2D), possibly because the vesicle connected to the plasma membrane through an exceedingly small (or transient) pore. An estimated 20% of all vesicles also have a similar behavior during normal release.
Interestingly, in the only preparation in which the fusion of the vesicle with the membrane can be directly followed, a different result was obtained: By imaging single vesicles of retinal bipolar cells of the goldfish with total internal reflection microscopy, Zenisek et al. (2002) did not detect a partial release of dye. On vesicle fusion, the dye spread laterally into the membrane within a few milliseconds, indicative of full fusion or of a large fusion pore that does not interfere with dye movements in the plane of the membrane (Fig. 2E).
- © 2012 Cold Spring Harbor Laboratory Press








