
Experimental Strategies for Cloning or Identifying Genes Encoding DNA-Binding Proteins
Adapted from Transcriptional Regulation in Eukaryotes: Concepts, Strategies, and Techniques, 2nd edition, by Michael F. Carey, Craig L. Peterson, and Stephen T. Smale. CSHL Press, Cold Spring Harbor, NY, USA, 2009.Abstract
This article describes experimental strategies for cloning or identifying genes encoding DNA-binding proteins. DNA-binding proteins are most commonly identified by electrophoretic mobility-shift assay (EMSA) or DNase I footprinting. To identify the gene encoding a protein detected by EMSA or DNase footprinting, the protein often needs to be purified and its sequence analyzed, as described here. Other methods are also available which do not resort to protein purification, including the one-hybrid screen, in vitro expression library screen, and mammalian expression cloning. These methods are outlined, and their advantages and disadvantages are discussed.
OVERVIEW
The most commonly used strategies for detecting proteins that bind a DNA element of interest are outlined in Experimental Strategies for the Identification of DNA-binding Proteins (Carey et al. 2012). If such activity is detected, experiments can be performed with antibodies to determine whether the protein corresponds to a protein predicted to bind the site in a TRANSFAC (http://www.gene-regulation.com/pub/databases.html) or JASPAR (http://jaspar.genereg.net) database analysis. Protein–DNA interaction assays can also be used to perform a basic characterization of the protein. For example, extracts from a number of different cell types or developmental stages can be analyzed to determine whether the binding activity is cell-specific or developmentally regulated. In addition, binding site mutants can be analyzed to identify the nucleotides required for the protein–DNA interaction. However, unless the protein is successfully identified by TRANSFAC or JASPAR, more advanced studies will depend on its identification using one of the strategies described below. After the protein and the gene encoding the protein are identified, their relevance for expression of the target gene can be assessed by gene disruption or RNA interference, the domains of the protein responsible for its activities can be determined by mutagenesis, and its mechanism of action and mode of regulation can be analyzed.
A preferred method for identifying a DNA-binding protein detected by EMSA or DNase I footprinting is to purify it and analyze proteolytic peptides derived from the pure protein by mass spectrometry. If the genome of the organism of interest has been fully sequenced and annotated, the peptide masses will likely match the masses of peptides expected from a protein encoded by an annotated gene, allowing the identification of the protein and its gene. This basic purification strategy is sometimes preferred because, following each purification step, the column fractions containing the binding activity can be monitored by EMSA or DNase I footprinting. The ability to follow the binding activity throughout the purification procedure ensures that the protein that is ultimately identified corresponds to the protein originally detected and characterized by EMSA or footprinting (Fig. 1). Another advantage of this approach is that binding activities that depend on two or more proteins or subunits can be purified; each subunit can then be identified by mass spectrometry. In contrast, most of the other methods described below will succeed only if the binding activity is a monomer, homodimer, or homomultimer.
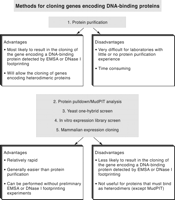
Methods for cloning genes encoding DNA-binding proteins.
The primary disadvantage of the protein purification strategy is that it can be challenging even for laboratories with protein purification experience and overwhelming for laboratories without it. A second limitation is that an abundant source of cells is sometimes required. However, as the sensitivity of mass spectrometry increases, the amount of starting material needed for successful purification and analysis is declining rapidly.
The other strategies are easy compared to the protein purification method, but they have a lower probability of yielding the gene encoding a binding protein observed in EMSA or DNase I footprinting. These strategies include one-hybrid screens, in vitro expression library screens, and mammalian expression cloning methods (Fig. 1), each outlined below. Protocols are not included because of space limitations, but appropriate references are provided. A common feature is that they do not rely on the initial development of a protein–DNA interaction assay. In other words, if an important DNA element has been identified in a promoter or distant control region, these techniques can be used to clone genes encoding proteins that specifically bind the element, without the need for preliminary EMSA or DNase I footprinting studies on crude extracts. Similarly, emerging strategies to identify proteins through simple pulldown experiments followed by multidimensional protein identification technology (MudPIT) analysis can also be pursued in the absence of an established protein–DNA interaction assay.
The identification of a DNA-binding protein and its gene in the absence of a preestablished protein–DNA interaction assay can be both an advantage and a disadvantage. Considerable time and effort can be saved if a protein–DNA interaction assay does not need to be developed and optimized. With a defined DNA element, cloning experiments can be initiated immediately. These strategies can also be used even if preliminary EMSA and DNase I footprinting experiments fail to detect a binding activity.
A principal disadvantage is that a gene and encoded protein that are isolated using a one-hybrid screen or expression screen possess a somewhat lower probability of being relevant for the function of the control element of interest. Furthermore, when using a MudPIT approach, a large number of candidate proteins will likely be identified that need to be evaluated to identify the small number of sequence-specific DNA-binding proteins that have the potential to be functionally relevant.
To explain why one-hybrid screens and expression screens are less likely to lead to the relevant protein, consider the following example of a DNA sequence element that has been carefully analyzed by mutagenesis in a functional assay. If an EMSA complex is detected using crude extracts, mutations in the control element can be analyzed to determine whether complex formation requires the functionally important nucleotides. A preliminary analysis of the expression pattern of the protein can also be performed to determine whether it corresponds to the anticipated pattern. If these preliminary studies provide compelling evidence that the protein is not relevant, the EMSA conditions can be varied until a more attractive candidate for the relevant DNA-binding protein is identified. One's ability to perform this preliminary characterization can greatly enhance the probability that the protein that is subsequently purified and cloned will correspond to the functionally relevant protein. In contrast, if a one-hybrid screen or expression screen is used, little evidence will be available before the screen is performed that the genes identified will encode DNA-binding proteins that recognize functionally relevant nucleotides and exhibit the anticipated expression pattern. This important information can be obtained only after the screen has been completed.
On the basis of the above considerations, the protein purification strategy is recommended if a particularly attractive candidate for a relevant DNA-binding protein has been identified by EMSA or DNase I footprinting, and if the laboratory has experience with protein purification procedures. In addition, the protein purification strategy may be essential if the protein–DNA interaction requires a heterodimeric or heteromultimeric protein. On the other hand, if an attractive candidate has not been identified by EMSA or footprinting, or if the laboratory has little experience with protein purification, the other approaches, in particular the one-hybrid screen or a simple pulldown approach followed by MudPIT analysis, are recommended as a starting point.
In the following sections, the principal strategies that are used to clone or identify genes encoding DNA-binding proteins are described (see Fig. 1). Protocols are not included because of space limitations. However, appropriate sources of detailed protocols are provided.
CLONING BY PROTEIN PURIFICATION AND PEPTIDE SEQUENCE ANALYSIS
The most successful strategy for purifying mammalian DNA-binding proteins uses column chromatography resins covalently linked to oligonucleotide multimers containing the DNA sequence of interest. This technique, known as sequence-specific DNA affinity chromatography, was developed by Kadonaga and Tjian to purify Sp1 (Kadonaga and Tjian 1986; Kadonaga 1991; Ausubel et al. 1994, Unit 12.10; Marshak et al. 1996). It has been used since to purify and obtain peptide sequences for more than 100 proteins (Marshak et al. 1996). A few mammalian DNA-binding proteins have been purified without resorting to DNA affinity chromatography (e.g., see Landschulz et al. 1988). However, because of the generally low abundance of mammalian transcription factors in cell extracts, purification by conventional methods is more difficult and rarely successful. Detailed descriptions of the method can be found in the references cited above. These references should be consulted for specific protocols and additional advice; central issues and supplementary suggestions are discussed here.
Amount of Starting Material
One important issue is the amount of starting material needed to purify and obtain peptide sequence or mass information for a mammalian DNA-binding protein. Before sensitive mass spectrometry techniques were developed, 50–100 pmoles of a purified protein was needed to obtain sufficient peptide sequence information for gene cloning by Edman degradation. Because of the low abundance of many sequence-specific DNA-binding proteins in nuclear extracts and the low yields obtained during DNA affinity chromatography, it was not unusual to need 100 L or more of cultured cells. Alternatively, animal tissues were a useful source when large quantities were required. The purification and cloning of NF-κB, for example, benefited greatly from the use of rabbit lung as a source of starting material (Ghosh et al. 1990). Unfortunately, many animal tissues contain high concentrations of proteases.
The emergence of increasingly sensitive mass spectrometry has greatly diminished the need for enormous quantities of the starting material. Less than 1 pmole of protein can now be detected readily by mass spectrometry. Therefore, the only requirement for analysis of a pure protein is the ability to detect the protein band on a stained SDS–PAGE gel for excision and further analysis. Even without a band on a stained gel or knowledge of the protein's molecular weight, mass spectrometry can be used to identify DNA-binding proteins that bind DNA affinity columns. The column eluate can be analyzed by MudPIT, or the eluate can be separated by SDS–PAGE, followed by dividing the entire gel lane into 10–20 slices. Each slice can then be analyzed by conventional mass spectrometry methods, such as Nano-LC/MS/MS (liquid chromatography/tandem mass spectrometry) (Domon and Aebersold 2006; Cravatt et al. 2007).
Conventional Chromatography Steps
Conventional chromatography steps are often included before the DNA affinity column. Gel filtration is often the most effective conventional step (Kadonaga and Tjian 1986; Kadonaga 1991; Marshak et al. 1996) because it separates proteins on the basis of a property (i.e., size) that is not linked to the DNA-binding activity of the protein. In other words, the protein of interest will be separated from most other nucleic-acid-binding proteins during gel-filtration chromatography. Gel filtration, however, requires a large column. Because of the challenge of pouring and running such columns, ion exchange columns using heparin or DEAE resins are more commonly used as a first step. Although these columns are easier to use and have been used successfully for initial fractionation (e.g., see Hahm et al. 1994), many DNA-binding proteins bind these resins with comparable affinities; therefore, they usually do not separate the protein of interest from the majority of other nucleic-acid-binding proteins in the extract. The number of conventional columns needed prior to affinity chromatography can vary. Some proteins, including Sp1, AP-1, and Ikaros, required only one gel filtration or ion exchange column before affinity chromatography (Kadonaga and Tjian 1986; Lee et al. 1987; Hahm et al. 1994). However, other proteins, including NF-κB, required several (Ghosh et al. 1990).
DNA Affinity Chromatography
The preparation and running of DNA affinity columns are described in detail in Kadonaga and Tjian (1986), Kadonaga (1991), Ausubel et al. (1994, Unit 12.10), and Marshak et al. (1996). Once the protein has been purified to a point at which (at most) a few abundant bands are observed on a silver-stained gel, each candidate band can be excised for mass spectrometry analysis, followed by an evaluation of each candidate protein.
The success of DNA affinity chromatography can be improved by generating multimers by polymerase chain reaction (PCR; Hemat and McEntee 1994). Traditionally, oligonucleotide monomers are annealed, phosphorylated, and ligated until long multimers are observed on an ethidium bromide-stained agarose gel (Kadonaga and Tjian 1986). Although this method has been used with considerable success, long multimers can be difficult to obtain for reasons that are not well understood. To prepare multimers by PCR, complementary oligonucleotides are synthesized that contain dimers of the binding site. During each PCR cycle, a fraction of the oligonucleotides will anneal in a staggered manner, allowing the gradual generation of long multimers.
DNA affinity chromatography procedures are similar to conventional chromatography procedures. One difference, however, is that the success of a DNA affinity step can usually be enhanced by overloading the column with protein. Whereas 10–40 mg of protein/mL is generally applied to a conventional chromatography column, 100 mg of protein/mL can be applied to a DNA affinity column. This can result in overloading, and a significant fraction of the protein of interest will not bind the column. However, overloading can saturate the column with the protein of interest, which increases the concentration of the protein in the high-salt eluates. The relatively higher concentration can contribute to the detection and stability of the protein. Although overloading greatly reduces the overall yield of pure protein, it may be necessary to purify the protein to a sufficient extent for analysis by mass spectrometry. In other words, yield may need to be sacrificed for purity.
Another difference between conventional chromatography and DNA affinity columns is that small-scale (i.e., pilot) DNA affinity columns are often unsuccessful. When developing a conventional chromatography procedure, it is quite common to establish conditions by loading relatively small amounts of protein onto a small column, in some cases less than the recommended amount per milliliter of resin. With DNA affinity columns, this is often unsuccessful because the protein within the eluates may not be stable or sufficiently concentrated for detection. If binding activity is not obtained during small-scale pilot experiments, it may be necessary to repeat the experiment on a larger scale.
Adding nonspecific competitor DNA to DNA affinity columns also sacrifices overall yield to achieve purity. Competitor DNA is usually preincubated with the protein sample before it is applied to the column. The procedure to determine the amount of competitor to add is described in Kadonaga (1991) and Marshak et al. (1996). The competitor binds to DNA-binding proteins that do not specifically bind the oligonucleotides linked to the column, thereby preventing them from efficiently binding the column. In practice, the competitor also prevents the protein of interest from binding the column to some extent. Reducing the amount of competitor can sometimes enhance the yield of the protein of interest. However, a high concentration of competitor may be required for optimal purity.
Confirmation That a Gene Identified Encodes the DNA-Binding Activity of Interest
To confirm that a gene identified following DNA affinity chromatography and mass spectrometry encodes the protein originally characterized by EMSA or DNase I footprinting, two experimental strategies are needed. First, the recombinant protein can be produced (by in vitro translation or expression in E. coli, HEK293T cells, etc.) and its DNA-binding properties compared with those of the protein detected in crude extracts. The recombinant protein should bind the same DNA sequence with the same nucleotide requirements as the protein that was purified. Furthermore, if the recombinant protein is full-length, it may generate an EMSA complex that comigrates with the complex detected in crude extracts, or it may generate a DNase I footprint or methylation interference pattern indistinguishable from that generated by the crude or purified protein. The second approach is to prepare or obtain antibodies against the recombinant protein, or against a synthetic peptide, and show that they react with the crude and purified protein. The antibodies should be capable of supershifting or disrupting the EMSA complex that was observed with the crude extract. If these experiments confirm that the gene encodes the protein of interest, one can assess the potential role of the encoded protein in the function of the control element to which it binds.
ALTERNATIVE CLONING METHODS
One-Hybrid Screen
The one-hybrid screen, typically performed in yeast, is sometimes an attractive method for identifying genes encoding proteins that bind a DNA element of interest (Fields and Song 1989; Li and Herskowitz 1993; Wang and Reed 1993; Inouye et al. 1994). A reporter plasmid is constructed that contains multiple copies of the binding site of interest upstream of a TATA box and a reporter gene (e.g., HIS3 or lacZ) (Fig. 2). This reporter plasmid is stably integrated into the yeast genome. Next, a library containing cDNAs from an appropriate cell source is prepared. The library is designed so that the encoded proteins are synthesized as fusions with a strong transcriptional activation domain. The yeast cells harboring the reporter gene are transformed with the library. Yeast cells expressing a fusion protein capable of binding the multimerized site express the reporter gene, and reporter expression is usually monitored by a selection strategy. For example, expression of a HIS3 reporter is monitored by the growth of cell colonies on minimal medium lacking histidine. Finally, the cDNA can be isolated from the selected cells and characterized further to confirm that the encoded protein binds the DNA sequence of interest.
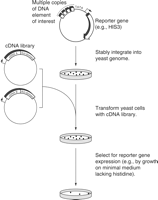
One-hybrid screening strategy.
The one-hybrid approach has been used to clone several important DNA-binding proteins and possesses several advantages. First, it is relatively straightforward and rapid. Second, the proteins are screened for binding under relatively native in vivo conditions. In contrast, protein purification and expression library screening (described below) depend on binding in vitro. Although mammalian proteins expressed in yeast are unlikely to acquire all of the posttranslational modifications found in mammalian cells, they may acquire one or more essential modifications. A final advantage is that the method is often extremely sensitive and can identify proteins that bind with only moderate affinity.
These advantages need to be balanced against two notable disadvantages: (1) The method will generally succeed only if binding requires a protein monomer, homodimer, or homomultimer and (2) the procedure will not necessarily identify the gene encoding a protein from a mammalian crude extract detected in an EMSA or DNase I footprinting assay. An additional caveat of the one-hybrid approach is that the binding site used must not interact with an endogenous yeast activator protein. If a yeast activator binds the site, reporter gene expression will be observed in the absence of the relevant cDNA expression plasmid. To overcome this, it may be possible to use variants of the binding site that retain activity in their natural context but do not bind yeast activators.
Despite its drawbacks, the one-hybrid procedure is strongly recommended as a starting point for gene identification if there is a reasonable chance the site can be recognized by a monomer, homodimer, or homomultimer. Detailed procedures for performing a one-hybrid screen can be found in the articles cited above or obtained from Clontech, which markets a kit for performing one-hybrid screens and preparing one-hybrid libraries (the Matchmaker Gold Yeast One-Hybrid Library Screening System). It is worth noting that the yeast one-hybrid assay has been adapted for use in high-throughput efforts to identify proteins capable of binding a collection of promoters, for the purpose of elucidating transcription regulatory networks (Deplancke et al. 2004, 2006).
In Vitro Expression Library Screening with DNA or Antibody Probes
Before the one-hybrid screening method was developed, a common method for cloning genes encoding novel DNA-binding proteins involved screening an expression library with radiolabeled DNA probes. This method (Singh et al. 1988) was an important advance in gene cloning technology. For a detailed description of the expression library screening method, see Ausubel et al. (1994, Unit 12.7). The technique requires a cDNA library prepared with mRNA from the cell type of interest. The library must be constructed in a λ bacteriophage vector that allows inducible expression of the protein encoded by the inserted cDNA after infection of E. coli. The recommended vector, λgt11, expresses proteins as β-galactosidase fusion proteins. Successful results have been obtained with libraries prepared by priming with either oligo(dT) or random oligonucleotides. The phage library is plated on bacteria under lytic growth conditions (Fig. 3). When plaques appear, transcription of the cDNA is induced by placing a nitrocellulose filter soaked in a solution of the inducing agent isopropyl-β-d-thiogalactoside on the plates. As the phage lyse, the induced proteins adhere to the nitrocellulose filters. After removal from the plates, the filters are incubated in a blocking solution to prevent nonspecific interactions between the probe and nitrocellulose filter. The filters are then probed with a radiolabeled DNA fragment containing multiple copies of the binding site of interest. The probe should bind specifically to plaques containing sequence-specific DNA-binding proteins. A nonspecific competitor DNA is included in this binding reaction to suppress nonspecific interactions by the radiolabeled probe. In some procedures (e.g., see Vinson et al. 1988), the expressed proteins on the filter are first subjected to a denaturation/renaturation procedure, with the aim of increasing the number of properly folded protein molecules capable of binding the probe. After the positive plaques are isolated and rescreened for the purpose of plaque purification, lysogen extracts can be prepared to allow the recombinant fusion protein to be characterized by EMSA.
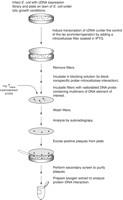
In vitro expression cloning.
Like the one-hybrid approach, this technique will not succeed if the protein must bind DNA as a heteromer and may not lead to the identification of the gene encoding the protein identified by EMSA or DNase I footprinting. In addition, it can be challenging to generate probe molecules with sufficiently high specific activity and a sufficient number of binding-site copies. In the original protocol, probes were prepared by excising from an appropriate plasmid a restriction fragment containing multiple copies of the binding site, followed by phosphorylation (Singh et al. 1988). The use of PCR to prepare long multimeric probes (as described above for preparing DNA affinity columns) can enhance the success of this approach (Hemat and McEntee 1994; Schmitt and McEntee 1996).
For most studies, the one-hybrid screen is more attractive than the in vitro expression library screen. The one-hybrid screen does not require the time-consuming phage titration steps generally required before an in vitro expression library screen can be performed. In addition, the one-hybrid screen does not require large amounts of radioactivity. Finally, the one-hybrid screen is less susceptible to protein folding and stability problems that can be encountered during the in vitro screen. The in vitro expression library screen is therefore recommended primarily if the one-hybrid screen and protein purification strategies fail to identify the relevant transcription factor.
Mammalian Expression Cloning Methods
Mammalian expression cloning methods have been used to identify new genes for three decades. Basically, a gene of interest is transferred to a recipient mammalian cell line, usually by transfection with sheared genomic DNA or a cDNA expression library. The cells that take up the gene of interest are identified by either selection or screening.
An early example of the use of a selection strategy for expression cloning was the identification of proto-oncogenes based on their ability to confer a transformed phenotype on recipient cells (see, e.g., Shih et al. 1979). Screening strategies have been used to isolate dozens of new genes, including several that encode cell surface proteins. One common screening procedure requires antibodies directed against the protein of interest and a cDNA expression library in a plasmid vector (see Seed 1987; Ausubel et al. 1994, Unit 6.11). The expression library contains the cDNAs downstream from a strong promoter and enhancer. The plasmids also contain an SV40 replication origin, which allows replication to high copy number in SV40 T-antigen-expressing COS-7 monkey kidney cells. The library is divided into several pools, each containing a diverse mixture of clones. The pools are then introduced into the COS-7 cells, resulting in plasmid replication and cDNA expression. To isolate a gene encoding a cell surface protein, each transfected cell population is analyzed for protein expression by panning. This involves immobilizing a specific antibody on a plastic dish, which facilitates the adherence of cells expressing the surface protein and the removal of nonexpressing cells. Plasmid DNAs from the adherent cells are isolated and amplified in E. coli. The resulting plasmids are again divided into pools and the entire procedure (i.e., transfection, panning, plasmid recovery, and amplification) is repeated several times, eventually leading to a single plasmid species containing the cDNA of interest.
Because of the success of the mammalian expression cloning strategies described above, several laboratories have attempted to use similar procedures to clone transcription factors that bind defined control elements or regions. For some experiments, synthetic promoters containing multiple copies of the site of interest were used to drive expression of a dominant selectable marker gene. In others, the gene encoding a cell surface protein was placed downstream from a synthetic promoter, with the expectation that the panning procedure could be used to isolate a cDNA encoding the relevant transcription factor. Native tissue-specific promoters were also tested; it was presumed that recipient cells would contain all of the ubiquitous factors needed for transcription, allowing the promoter to function when a cDNA encoding an essential tissue-specific factor was provided. Unfortunately, these strategies rarely were successful. Their failure was likely the result of a combination of factors, particularly the inability of a single transcription factor to induce transcription to a sufficient extent above background.
Despite these shortcomings, mammalian expression cloning methods occasionally have been used to clone genes encoding proteins important for gene regulation. One example is the cloning of the gene encoding SCAP (SREBP cleavage-activating protein; Hua et al. 1996), which regulates the function of SREBP-1, the sterol response element binding protein (Yokoyama et al. 1993). To clone SCAP, a plasmid cDNA library was transfected into human 293 cells in pools of about 1000 clones, along with a plasmid containing a luciferase reporter gene under the control of multiple SREBP-1-binding sites. The library was prepared from mRNA from sterol-resistant cells, and the goal of the study was to isolate the gene responsible for sterol resistance. In response to sterol overload, normal cells down-regulate the expression of SREBP-1-responsive genes. Thus, cells that express the protein that confers sterol resistance should show higher luciferase activity than normal 293 cells in the presence of high concentrations of sterol. Indeed, a plasmid pool was identified that yielded high luciferase activity. Sequential analysis of plasmid subpools led to the cloning of the SCAP gene. Functional analysis of SCAP revealed that it cleaves membrane-bound SREBP-1, allowing it to translocate to the nucleus and activate transcription. This example raises the possibility that improvements in the technology for preparing and analyzing plasmid libraries will allow the cloning of other transcriptional regulators by similar methods.
- © 2012 Cold Spring Harbor Laboratory Press








