
Imaging Microglia in Brain Slices and Slice Cultures
Adapted from Imaging in Neuroscience (ed. Helmchen and Konnerth). CSHL Press, Cold Spring Harbor, NY, USA, 2011.Abstract
Here we describe a method for imaging fluorescently labeled parenchymal microglia (MG) in excised neonatal or adult rodent brain tissue slices. Using multichannel confocal or two-photon time-lapse imaging, the approach affords real-time analyses of MG behaviors, including motility, migration, chemotaxis, proliferation, and phagocytosis in live brain tissues. The method is applicable to acutely prepared tissue slices from developing and adult rodents and to slice cultures derived from neonatal rodents, including transgenic and green fluorescent protein reporter mice. A variety of fluorescent tags can be used to study the structure and physiology of MG in these preparations. Moreover, bath application of reagents (such as ATP) can establish spatial and temporal gradients that induce chemokinesis- and chemotaxis-like MG migration in tissue slices. Thus, the approach is useful for dissecting the molecular basis of MG behaviors and testing whether candidate reagents alter MG behavior and function in semi-intact central nervous system tissue preparations.
MATERIALS
Reagents
Dissection medium (DM)
-
Prepare this medium by supplementing Hanks’ balanced salt solution (HBSS) with 6 mg/mL dextrose.
Filter culture medium (FCM)
-
This medium contains 50% modified Eagle’s medium (MEM), 25% HBSS, 25% horse serum, 2 mm glutamine, 0.044% NaHCO3, and 10 units/mL penicillin–streptomycin.
HEPES-buffered culture medium (HCM)
-
This medium contains 50% MEM, 25% HBSS, 25% horse serum, 6 mg/mL d-glucose, and 25 mm HEPES.
Isolectin B4 (IB4 or GS-IB4) derived from Griffonia simplicifolia seeds
-
Several fluorescent IB4 conjugates are commercially available (FITC-conjugated IB4 from Sigma-Aldrich; and Alexa Fluor 488-, 568-, 594-, or 647-conjugated GS-IB4 from Life Technologies). They all yield strong microglial staining and serve as excellent markers for resting or activated parenchymal MG in rodent brain tissues (Streit and Kreutzberg 1987). The Alexa Fluor 647 fluorophore is especially suitable for work in thick tissue slices because longer wavelength excitation and emission light show less tissue scatter. IB4 labeling can be performed by diluting the lectin to 5 µg/mL in any of a variety of solutions, including FCM, HCM, MBSS, or phosphate-buffered saline.
Live- or dead-cell fluorescent markers (e.g., Sytox Orange, TO-PRO-3, YO-PRO-1)
Modified balanced salt solution (MBSS)
pHrodo-labeled Escherichia coli bioparticles
Rodents of experimental interest
-
Such animals may include Sprague–Dawley rats, C57BL/6 mice (Harlan Laboratories), and green fluorescent protein (GFP) and yellow fluorescent protein (YFP) reporter mouse lines (Jackson Laboratory).
Vacuum grease
Equipment
Coverslips
Imaging chamber (made from Plexiglas; see Step 3)
Imaging setup
-
For imaging MG in thick tissue slices and slice cultures (50–500 µm thick), we recommend optical sectioning with a confocal or two-photon imaging system. We use commercially available laser-scanning confocal or two-photon microscopes equipped with multiple lasers, computer-driven focus, and automated image capture (e.g., Leica SP2 AOBS confocal imaging system on an inverted microscope platform or a Leica SP5 two-photon system on an upright platform). Use the manufacturer’s software for controlling the microscope and for image capture.
Imaging software (Photoshop or ImageJ)
Microscope stage heater
-
Stage heating can also be accomplished with a simple, custom-made heater modified from a commercially available hair dryer.
Nylon mesh (110-μm window, 47% open area) (Small Parts, Inc., CMN-0112-C)
Objective lens (20×, 0.7-NA, dry Plan-Apochromat)
Syringe (1-mL)
Tissue culture dishes (six-well)
Tissue culture incubator (5% CO2, 36°C, 100% humidity)
Tissue slicer (manual) (Stoelting Co. 51425)
METHOD
Preparation of Tissue Slices from Rodent Brain
-
This procedure should take 5–15 min.
-
1. Prepare tissue slices using one of the following methods.
-
For immediate imaging: Prepare acutely isolated hippocampal tissue slices from neonatal (postnatal day [PND] 5–7) or adult (6- to 8-wk-old) Sprague–Dawley rats or C57BL/6 mice according to the method described in Dailey and Waite (1999). Decapitate the animal. Remove the brain and place it in cold DM. Excise the hippocampus and section it transversely to obtain 300- to 400-μm-thick slices using a manual tissue slicer.
-
For organotypic slice cultures and later imaging: Prepare slices from neonatal (PND 5–7) animals using the static filter culture method originally described by Stoppini et al. (1991). Place the slices on porous membrane inserts in six-well plates. Incubate in FCM in a humidified tissue culture incubator (5% CO2 and 36°C).
-
A more detailed protocol for the preparation of rodent hippocampal slices and slice cultures is available elsewhere (Fuller and Dailey 2007).
-
-
Labeling of Microglia in Tissue Slices with Fluorescent Lectin
-
This procedure should take 45–60 min.
-
2. Label MG in neonatal tissue slices and organotypic slice cultures by incubating in HCM (ambient air) or FCM (5% CO2 environment) containing 5 µg/mL of fluorescently conjugated IB4 lectin for 45–60 min at 36°C. Label MG in hippocampal slices from mature rodents by incubating in MBSS containing 5 µg/mL IB4 under the same conditions. For the free-floating tissue slices, incubate in a 35-mm dish or multiwell plate and then rinse with dye-free medium for 5–10 min. For the organotypic slice cultures, use a scalpel to cut out sections of membrane with attached slices and incubate these membrane-attached tissue slices in a dish with HCM containing IB4. Make sure all slices are submerged and do not dry out.
-
As an alternative to lectin labeling, live slices or slice cultures can be prepared from heterozygous mice expressing GFP in MG. GFP expression levels vary somewhat with MG activation state. However, GFP expression (in MG from Cx3cr1 mice) is strong enough for good visualization of both activated and quiescent MG in living or formaldehyde-fixed tissue slices.
-
Lectin labeling (or GFP expression) can be combined with other fluorescent labeling techniques to evaluate phagocytosis of dead or dying cells (nuclei of compromised cells labeled with Sytox Orange or TO-PRO-3) or bacterially derived bioparticles (pHrodo-labeled E. coli bioparticles). Alternatively, reporter lines expressing GFP in MG can be crossed to available lines expressing YFP in neurons (e.g., Thy1-YFP).
-
Mounting Tissue Slices for Imaging
-
This procedure should take 5–10 min. Methods for mounting slices for imaging vary with the tissue preparation. It is important to secure the tissue slices to prevent movement during imaging while retaining sufficient gas exchange and access to media. Procedures for mounting acutely prepared slices are given in Steps 3–7. Details for mounting organotypic slice cultures affixed to membranes are described in Dailey et al. (2011).
-
3. To mount acutely prepared (free-floating) tissue slices from neonatal rodents, construct a simple imaging chamber from a custom-made Plexiglas slide (75 × 25 × 3-mm) with an oblong hole (35 × 13-mm) cut out in the center (Dailey et al. 2011). Form the chamber floor by securing a 50-mm glass coverslip with vacuum grease to the bottom of the Plexiglas slide under the hole. Apply two thin lines of vacuum grease onto the coverslip floor (∼5 mm long and ∼5 mm apart) using a “grease gun” custom made from a 1-mL syringe with a flame-tapered tip.
-
4. Transfer one to three tissue slices with medium to the chamber and position the slices carefully between the lines of vacuum grease.
-
5. Lay a piece of nylon mesh over the slices and the grease lines, then gently press it into the grease with forceps to secure the slices in place under the mesh. Take care not to press too firmly as this will compress the tissue and inhibit MG cell movements.
-
6. (Optional) To increase the chamber volume, place a second Plexiglas slide on top of the first slide and align the holes. Secure with vacuum grease.
-
7. Add medium to cover the tissue slices. Lay a second coverslip over the top of the chamber to prevent evaporation.
-
Focal drift can be reduced if the chamber is not completely filled with medium (i.e., leave an air pocket in the chamber so that the top coverslip is not tightly sealed).
-
Confocal and Two-Photon Imaging
-
This procedure should take 30 min–24 h.
-
8. To capture a large field of view (∼500 × 500-μm) containing several (typically 10–30) MG cells, collect images using a 20×, 0.7-NA dry Plan-Apochromat objective lens.
-
9. Capture images using a confocal or two-photon microscope.
-
To view the dynamic activities of MG in live tissues, capture images at intervals of a few seconds to minutes, over total periods of tens of minutes to several hours. A 20×, 0.7-NA dry objective lens is sufficient to observe many dynamic activities of MG for several hours at a time, at submicron pixel resolution, with an incident illumination power of <20 µW at the specimen. To monitor cell movements in three dimensions using a 20× objective lens, collect optical sections at 2–3 µm z-step intervals over 30–40 µm of tissue depth at time intervals of 3–4 min. Capture images more frequently for higher time resolution.
-
-
10. Process and analyze images using Photoshop or ImageJ.
-
For playback and analysis, compile image stacks and combine into extended focus time-series images using the microscope manufacturer’s software or ImageJ. Because some lateral x–y drift of the field of view is common, it is very useful to perform postprocessing registration of images in a time series using ImageJ plug-ins (e.g., Stackreg). Time stamps and scale bars are also easily added to the time series using ImageJ plug-ins. Playback the time series in ImageJ using the stack tools or export from ImageJ for playback in QuickTime.
-
See Troubleshooting.
-
TROUBLESHOOTING
Problem (Step 10): IB4 labeling of MG appears weak relative to labeled blood vessels.
Solution: The IB4 lectin labels endothelial cells of blood vessels in addition to MG, and lectin staining of MG varies somewhat with species and developmental age. IB4 lectin staining is stronger in tissues from rat than from mouse. In mouse tissues, lectin staining is stronger in tissues from developing mice than from adult mice. However, in fixed tissues, IB4 lectin staining can be significantly improved in mouse tissues if they are fixed with methanol as opposed to formaldehyde. Finally, IB4 lectin becomes internalized in active MG, so visibility of the full MG cell structure is somewhat reduced after 10 h. Mouse lines in which GFP is expressed in the microglia (Jung et al. 2000; Hirasawa et al. 2005) provide superior visibility, but they are less readily available.
Problem (Step 10): MG movements appear sluggish in time-lapse movies.
Solution: MG motility and migration in live tissue slices from developing and mature rodents are sensitive to temperature. Stage heating (preferably above 32°C) is necessary for maximal movement. Although MG in tissues from neonatal rodents are relatively resistant to hypoxia, MG in tissue slices from mature rodents are sensitive to oxygen levels. Adult tissues require oxygenated medium. Finally, MG movements appear to be hindered if tissue slices are mechanically compressed during mounting.
DISCUSSION
The methods described here should yield highly visible MG that show spontaneous movements and respond rapidly to exogenously applied reagents. Cells and tissues should be viable for hours without evidence of photobleaching or rundown induced by poor chamber conditions or phototoxicity.
Using this approach, we have shown that MG undergo rapid changes in cell morphology and motility following activation in live rodent brain tissues (Stence et al. 2001). Within minutes, MG in acutely isolated hippocampal tissue slices begin to activate and subsequently accumulate near injured pyramidal neurons, a process we refer to as “homing” (Kurpius et al. 2006). MG show a chemotaxis-like response to exogenously applied purines, including ATP and ADP (see Fig. 1 and Kurpius et al. 2007), and studies in tissues from knockout mice have shown that this is dependent on P2Y12-type purinoceptors (Haynes et al. 2006). Moreover, use of cultured tissue slices has shown that MG remain active for several days after tissue injury and play important roles in phagocytic uptake and clearance of dead cell debris (Petersen and Dailey 2004). Thus, the combination of acutely excised rodent tissue slices and cultured tissue slices has provided excellent ex vivo models for studying MG activation and motility in a central nervous system (CNS) tissue environment.
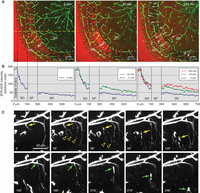
Migration of microglia (MG) in rat and mouse tissue slices following application of exogenous ADP. (A,B) A rat hippocampal slice was stained with Alexa Fluor 488-IB4 (green) and a mixture of ADP (1 mm) and fluorescently tagged ADP (BodipyTR-ADP, 10 µm; red) was added to the bathing medium. Selected images from a time-lapse confocal imaging session show diffusion of BodipyTR-ADP into the slice. Scale bars, 50 µm. (B) Measurement of BodipyTR-ADP fluorescence across the dashed yellow line for each of the three time points shown in (A). Note the steep concentration gradient across the stratum orients (SO). Levels of BodipyTR-ADP continue to rise in the interior of the slice (stratum radiatum, SR) over the 3-h time period imaged. During this time, many MG cells migrated up the gradient toward the edge of the slice. Arrows indicate the net movement of individual MG over the 3-h period. See Movie 1. (C) Time-lapse sequence in a different slice showing MG branch extension followed by cell migration in response to bath application of exogenous ADP (1 mm). ADP was applied to the slice immediately after excision, and imaging commenced 1 h later. When ADP is first applied at early stages of activation (as here), MG (arrows) first extend long branches (open arrowheads) directed toward the source of ADP, followed by MG cell migration. Two migrating MG are highlighted (yellow and green arrows, respectively) in this 4-h time sequence. See Movie 2.
Migration of microglia (MG) cells in a rat hippocampal tissue slice following bath application of exogenous adenosine diphosphate (ADP) (1 mm). The red fluorescence is BodipyTR-ADP.
Branch extension and subsequent migration of IB4 lectin-labeled microglia in rat hippocampal tissue slices following application of exogenous adenosine diphosphate (ADP) (1 mm).
When GFP-expressing tissues are unavailable or undesirable, fluorescent conjugates of IB4 lectin provide a simple, one-step method for labeling MG. IB4 penetrates tissues efficiently and selectively stains MG and endothelial cells lining blood vessels by binding to terminal α-d-galactosyl residues of glycoproteins and glycolipids on these cells (Streit and Kreutzberg 1987). This selective surface staining provides good structural definition of MG. The variety of fluorescent IB4 conjugates allows flexibility for double or triple labeling with other fluorescent markers, including markers of live or dead cell nuclei (see Fig. 2 and Dailey and Waite 1999).
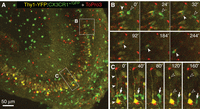
Three-channel, time-lapse confocal imaging in a live tissue slice from a double-reporter mouse expressing GFP in MG (green) and YFP in pyramidal neurons (yellow). The slice was counterstained with TO-PRO-3 to label nuclei of dead cells (red). (A) Low-magnification view of hippocampal area CA3. (B) Selected higher magnification images from a time-lapse series shows movement of two MG cells (arrowheads), one (solid arrowhead) containing engulfed TO-PRO-3-labeled dead cell nuclei. (C) A GFP-expressing MG cell (arrow) passes by a YFP-expressing pyramidal neuron with labeled cell body and apical dendrite (solid arrowheads). Several hours after injury, the neuron shows signs of degeneration, evident as dendrite blebs (open arrowheads at 120′ and 160′). The present methods enable dynamic analysis of cell interactions between MG and other cells in central nervous system tissues.
Because IB4 is nontoxic to live cells, it is amenable to time-lapse imaging in living tissues. However, some limitations must be considered. First, it is essential to reduce illumination intensity to minimize phototoxic damage or bleaching. Fortunately, IB4-labeled MG are relatively hardy and can be subjected to confocal or two-photon imaging conditions over many hours without ill effects. Second, membrane turnover in activated MG reduces surface labeling over the course of a lengthy time-lapse imaging session (>10 h). This can result in a reduced fluorescent signal and apparent “fading” of the MG over the course of time. Finally, imaging MG in adult tissues is more challenging because, unlike neonatal tissues, the adult tissues require continuous perfusion with oxygenated media, and they are prone to showing signs of reduced movement under hypoxic conditions. Moreover, IB4 lectin staining of MG is weaker in adult mouse tissues in comparison to younger mouse or rat tissues. Some of these limitations can be overcome by using GFP reporter mouse lines (see Fig. 2). In summary, the present methods afford opportunities to study dynamic MG cell events as they occur in a brain tissue environment on a timescale of minutes, hours, and even days.
ACKNOWLEDGMENTS
This work was supported by grants to MED from the National Institutes of Health (NS-43468, AA018823), the Roy J. Carver Charitable Trust, and the American Heart Association (0950160G).
- © 2013 Cold Spring Harbor Laboratory Press








