
Purification of Astrocytes from Transgenic Rodents by Fluorescence-Activated Cell Sorting
Abstract
The purification of astrocytes by fluorescence-activated cell sorting (FACS) requires that an astrocyte-specific promoter drive the expression of the green fluorescent protein (GFP). Our laboratory uses FACS to acutely isolate astrocytes from young and old tissue as well as to isolate GFP-negative neurons at the end of the FACS sorting to conduct comparative unbiased, large-scale gene expression studies. Because of the relatively harsh nature of FACS sorting, few astrocytes or neurons survive long enough after the sort to be cultured.
MATERIALS
Reagents
BSA (4%)
-
To prepare a stock of 4% BSA in Dulbecco’s phosphate-buffered saline (D-PBS), dissolve 8 g of BSA (Sigma-Aldrich A4161) in 150 mL of D-PBS (HyClone SH30264.01) at 37°C. Adjust the pH to 7.4 with ∼1 mL of 1 n NaOH. Bring the volume to 200 mL. Filter through a 0.22-µm filter. Store in 1-mL aliquots at −20°C.
DNase (0.4%)
-
To prepare a 0.4% stock of DNase in Earle’s balanced salt solution (EBSS), add 1 mL of EBSS (Sigma-Aldrich E6267) per 12,500 units of DNase (Worthington LS002007 or Sigma-Aldrich D4527). Keep on ice. Filter-sterilize, and store in 200-µL aliquots at −20°C.
Dulbecco’s phosphate-buffered saline (D-PBS) containing Mg2+ and Ca2+ (HyClone SH30264.01)
-
Store at room temperature.
High-ovomucoid (high-ovo) stock solution (10×)
l-cysteine hydrochloride monohydrate (Sigma-Aldrich C7880)
IP-astrocyte base medium containing 5 ng/mL HBEGF (Sigma-Aldrich 4643)
-
Equilibrate all media as in Step 2.ii before use.
Low-ovomucoid (low-ovo) stock solution (10×)
Papain (Worthington LS003126)
PBS (Diamedix 1000-3)
Primary antibodies:
-
Anti-PDGFRα (rat IgG, monoclonal antibody CD140a clone APA5; BD Biosciences-Pharmingen)
-
O4 hybridoma supernatant (mouse IgM; Bansal and Pfeiffer 1989)
-
Galactocerebroside (GalC) supernatant (for use with mice older than P8)
-
Myelin oligodendrocyte glycoprotein (MOG) supernatant (for use with mice older than P8)
-
O1 supernatant (for use with mice older than P8)
Propidium iodide (1 mg/mL)
Secondary antibodies:
-
Goat anti-mouse IgG + IgM (H + L) (Jackson ImmunoResearch 115-005-044)
-
Goat anti-mouse IgM μ-chain specific (Jackson ImmunoResearch 115-005-020)
-
Goat anti-rat IgG (H + L) (Jackson ImmunoResearch 112-005-167)
-
Donkey anti-mouse APC (eBioscience 17-4012-82) (for use with mice older than P8)
Transgenic mice, 8–10 S100β-eGFP (P0–P30) or Aldh1L1-eGFP
Tris-HCl (50 mm, pH 9.5), sterilized
Trypan blue
Equipment
ACLAR plastic coverslips (ethanol-washed and PDL-coated) or tissue culture plates (15-cm; PDL-coated)
-
To coat tissue culture plastic with poly-d-lysine (PDL), dilute a 100× PDL stock solution (1 mg/mL in H2O) to 1× in sterile H2O, and then coat the plastic for at least 30 min. Wash three times with sterile H2O before drying or using. In practice, we coat ethanol-washed ACLAR coverslips after adding them individually to each well of a 24-well plate (see Step 59.i). ACLAR is very hydrophobic; add 500 µL of PDL to cover each coverslip completely. If the coverslip floats up, tap it down with a pipette tip. Wash the coverslips in the wells three times with sterile H2O before adding medium.
Aspirator
Bunsen burner and ethanol
Conical tubes (50-mL)
Dissection equipment:
-
Decapitation scissors, large (ROBOZ RS-6820)
-
Dissecting microscope
-
Dissection scissors, curved (ROBOZ RS-5675)
-
Forceps, #5
-
Forceps, curved
-
Scalpel with No. 10 scalpel blade
-
Scissors, medium-sized (ROBOZ RS-6702)
-
Scoop, perforated (Moria MC17)
Equilibration setup (a source of carbon dioxide [5% CO2/95% O2] with a line leading to a sterile hood)
FACS machine
Forceps
Heat block preset to 34°C in a sterile hood
Hemacytometer
Hood for sterile tissue culture
Incubator preset to 37°C and 10% CO2
Microscope, phase contrast
Nitex mesh filter (Tetko HC3-20)
-
Cut into 3-inch squares, wrap in small packets of foil, and autoclave.
Petri dishes, plastic (6-cm, 15-cm)
Petri dish lids (6 cm) with a hole in the center that accommodates a 0.22-µm filter
-
Use flamed forceps (spray with ethanol and use a Bunsen flame to sterilize) to melt the hole into the center of the lid.
Pipettes, serological (1-mL, 2-mL, 5-mL, 10-mL)
Pipettor, powered (e.g., Pipet-Aid)
Refrigerator preset to 4°C
Syringe, 20 mL
Syringe filters (0.22-µm)
Tabletop centrifuge (with 15 mL/50 mL centrifuge tube adaptors) at room temperature
Tissue culture plates, multiwell (24-well)
Water bath preset to 34°C
METHOD
-
In the method described here, brain tissue is dissected from transgenic rodents, a single-cell suspension is prepared, and cells are immunopanned before proceeding to FACS (see Fig. 1). Cells from animals older than P8 undergo additional steps before FACS to further deplete oligodendrocytes and myelin. The entire protocol is a 2-d procedure; only Step 1 is performed on the first day.
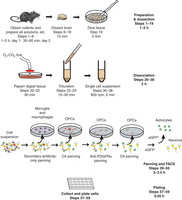
Purification of astrocytes from S100β–eGFP mice by fluorescence-activated cell sorting (FACS).
Preparation of Panning Dishes
-
1. Prepare panning dishes by coating four 15-cm Petri dishes with 25 mL of 50 mm Tris–HCl (pH 9.5) per dish and the appropriate secondary antibody solution.
-
One “secondary antibody only” dish: 60 µL anti-mouse IgG + IgM (H + L)
-
Two “O4” dishes: 60 µL anti-mouse IgM μ-chain specific
-
One “PDGFRα” dish: 60 µL anti-rat IgG
Incubate the dishes overnight at 4°C.
-
Plates are initially hydrophobic, but after coating overnight, they become visibly hydrophilic. If plates are needed immediately, a quick but less preferable way to make plates is to coat with secondary antibodies for 2 h at 37°C.
-
Preparation of Solutions and Panning Dishes
-
2. Aliquot and equilibrate the enzyme stock solution.
-
i. Aliquot 22 mL of enzyme stock solution into a 50-mL conical tube.
-
ii. Break a 2-mL pipette, attach a 0.22-μm filter, and attach it to the 5% CO2/95% O2 line. Bubble 5% CO2/95% O2 through the solution until it turns from red to orange (see Movie 1).
-
iii. Put the tube of equilibrated enzyme stock solution into a 34°C water bath.
-
Astrocytes are exquisitely sensitive to pH changes. Yield and viability are highly affected by improper equilibration of the enzyme and dissociation media. All media in contact with cells must be properly equilibrated before use, including the growth medium that will be added.
-
-
-
3. Aliquot and equilibrate the inhibitor stock solution.
-
i. Prepare two tubes with 21 mL per tube, and one tube with 10 mL.
-
ii. Bubble CO2 through the solution until it turns from red to orange.
-
The 2-mL pipette from Step 2 can be re-used for this step. Do not put the equilibrated inhibitor stock into the water bath.
-
-
-
4. Prepare 60 mL of 0.2% BSA/DNase: Combine 57 mL of DPBS with 3 mL of 4% BSA and 24 µL of 0.4% DNase.
-
5. Prepare 50 mL of 0.02% BSA/DNase: Combine 45 mL of DPBS with 5 mL of 0.2% BSA/DNase and 50 µL of 0.4% DNase.
-
6. Prepare high- and low-ovo inhibitor solutions.
-
i. Combine the following for the high-ovo inhibitor solution: 10 mL inhibitor stock solution (from Step 3) + 2 mL 10× high-ovomucoid stock solution + 20 µL 0.4% DNase.
-
ii. Combine the following for the low-ovo inhibitor solution (prepare two conical tubes): 21 mL inhibitor stock solution (from Step 3) + 1.5 mL 10× low-ovomucoid stock solution + 100 µL 0.4% DNase.
-
-
7. Finish preparing the panning dishes from Step 1.
-
i. Wash each dish three times with PBS.
-
ii. Rinse each dish again with sterile PBS, and then coat them with the appropriate primary antibody solutions. Swirl the dishes for even coating, and allow them to incubate with primary antibody for at least 2 h at room temperature before use in Step 40.
-
One “secondary antibody only” dish: 12 mL of 0.2% BSA/DNase
-
Two “O4” dishes: 8 mL of 0.2% BSA/DNase + 4 mL of O4 hybridoma supernatant
-
One “PDGFRα” dish: 12 mL of 0.2% BSA/DNase + 20 µL of PDGFRα antibody
-
-
-
8. After the 22 mL of enzyme stock solution from Step 2 has been warmed at 34°C for 10 min, add 100 units of papain and 3.2–4.0 mg (for 1 mm) l-cysteine-HCl. Swirl to ensure solvation. Keep the mixture warm in a 34°C water bath before beginning the dissection, but be sure to use it within 20 min of adding the papain.
-
Pre-activation of papain before usage optimizes the enzymatic activity, enabling shorter enzymatic dissociation times. More papain should be used for tissue older than P7. The amount should be increased according to the age of the brain. Typically, we would use 200 units for P14 brains and up to 420 units for P30 brains. For brains older than P14, use no more than four brains per preparation to avoid overloading the panning dishes and FACS machine.
-
Gas tank setup. Illustrates Steps 2 and 3.
Brain Dissection
-
Dissection should only take ∼20 min (see Movie 2).
-
9. Prepare two 6-cm Petri dishes with 10 mL of D-PBS per dish.
-
10. Grab the animal to be dissected by the scruff and swiftly decapitate it with scissors. Discard the body.
-
11. Insert medium-sized scissors under the skin of the animal’s head, avoiding the skull. Slide the scissors down the middle of the head to slice the skin into two halves. Gently pull apart the skin to reveal the skull.
-
12. Insert curved scissors into the base of the spinal cord, and make two cuts on each side. On one side, cut around the brain and horizontally across the part of the skull that covers the olfactory bulb.
-
13. Remove the skull flap, and cut the olfactory bulb with curved scissors.
-
14. Gently insert curved forceps under the brain. Cut the optic nerve and transfer the brain into one of the D-PBS-containing Petri dishes from Step 9. As more animals are dissected, split the brains evenly between the two dishes. Make sure that the brains are completely covered by D-PBS.
-
15. Under a dissecting microscope, removing the midbrain, hindbrain, and striatal regions, leaving only the cortex.
-
16. Remove the meninges from the surface of the cortex by peeling them off with forceps.
-
This is easier to do with younger animals but can be done until ∼P7 on mouse. Be careful not to make little tears in the surface of the cortex, so the meninges can be peeled off in sheets rather than in little sections.
-
-
17. Add a 200-μL drop of D-PBS to the center of two 6-cm Petri dishes.
-
18. Use a perforated scoop to transfer the cortices to the drops of D-PBS. Split the cortices evenly between the dishes. Put a maximum of five mouse cortices per Petri dish, to enable more thorough enzymatic dissociation (see Movie 3).
-
19. Use a No.10 scalpel blade to dice the brains into ∼1 mm3 pieces.
Brain dissection. Illustrates Steps 11–16.
Dissection of brains with scalpel. Illustrates Steps 17–22.
Dissociation of the Cells
-
After papain treatment to loosen contacts in the extracellular matrix, the tissue is washed and then mechanically dissociated by gentle sequential trituration using a 5-mL pipette with fresh inhibitor solution to yield a suspension of single cells. This dissociation needs to be quick yet gentle to tease the cells apart rather than rip them apart; ripping will result in poor cell health.
-
20. In a sterile hood, attach a 0.22-μm filter to a 20-mL syringe. Filter and discard 2 mL of the papain/enzyme stock solution prepared in Step 8. Then filter 10 mL of enzyme stock solution into each of the two Petri dishes containing finely diced brains (see Movie 3).
-
21. Add 50 µL of 0.4% DNase to each Petri dish and swirl the dishes.
-
22. Allow the brains to digest by leaving the dishes on a heat block set to 34°C. Cover with lids that have holes in the top to accommodate a 0.22-μm filter attached to the 5% CO2/95% O2 line. Blow CO2 over the brains continuously and swirl the dishes every 10 min (see Movie 3). For P8 and younger brains, leave the dishes for 80 min. For brains older than P8, leave for 90 min.
-
Avoid creating bubbles.
-
-
23. Transfer the digested cortices from both Petri dishes into a 50-mL conical tube. Wait for the tissue to settle, then aspirate and discard the excess liquid with a suction pump.
-
24. Wash the cells by adding 4 mL of low-ovo inhibitor solution from one of the tubes (“Tube 1”) prepared in Step 6.ii. Wait for the cells to settle, and then aspirate and discard the excess liquid.
-
25. Repeat Step 24 three times. There should be ∼5 mL of low-ovo inhibitor solution remaining in Tube 1. This is the low-ovo inhibitor solution to which single cells will be added in Step 28.
-
26. Add 4 mL of low-ovo inhibitor solution from the second tube prepared in Step 6.ii (“Tube 2”) to the conical tube of cells for trituration.
-
27. Use a 5-mL serological pipette to quickly suck up and release the solution of brain + low-ovo inhibitor solution (see Movie 4). As the tissue dissociates, the low-ovo inhibitor solution in the tube will become cloudy. Be careful not to introduce bubbles, and do not lift the 5-mL pipette out of the solution.
-
The dissociation buffer (the low-ovo inhibitor solution) contains Earle’s balanced salts, a bicarbonate-based buffer that requires careful equilibration with 5% CO2/95% O2 gas before use and during papain treatment. When the dissociation buffer is exposed to room air during trituration, minimizing surface area and avoiding bubbles is essential for maintaining the proper pH and cell health. When first using this procedure, poor cell health and viability is common, because it is hard to work quickly and efficiently. As the user becomes more practiced, an increase in cell health should be seen.
-
-
28. Allow the tissue to settle. A cloudy, single-cell suspension will form above the chunks of undissociated brain. Collect the single cells with a 1-mL pipette and add them to the 5 mL of fresh low-ovo inhibitor solution left in Tube 1 from Step 25.
-
29. Repeat the trituration (Steps 26–28) four times, until almost all of the brain chunks have dissociated into single cells.
-
Generally, 95% of the chunks are gone at the end of this step. Discard the remaining brain chunks, because their dissociation will not yield healthy cells.
-
-
30. Count the cells.
-
i. Dilute the cells 1:5 by combining 20 µL of cells with 80 µL of 0.02% BSA/DNase.
-
ii. Dilute this mixture 1:2 with trypan blue and count the cells using a hemacytometer.
-
The cells should look healthy under the phase microscope and have a viability exceeding 90% (measured by trypan blue exclusion).
-
-
-
31. Use a 1-mL pipette to remove any remaining undissociated brain chunks that have settled to the bottom of the single-cell suspension.
-
32. Use a 10-mL pipette to carefully layer 12 mL of high-ovo inhibitor solution under the single-cell suspension (see Movie 4).
-
High-ovo inhibitor solution is more dense than low-ovo inhibitor solution and will remain at the bottom, as long as its release is carefully controlled.
-
-
33. Centrifuge the cells at 110g for 5 min in a tabletop centrifuge at room temperature.
-
The idea here is for the dissociated cells to move down through the high-ovo inhibitor solution to ensure the complete inhibition of the papain.
-
Not all of the cells will come down at 110g, but viability is decreased at higher centrifugation speeds.
-
-
34. Aspirate and discard the supernatant. There should be a visible pellet of cells at the bottom of the conical tube.
-
35. Resuspend the cell pellet.
-
i. Add 3 mL of 0.02% BSA/DNase, and gently resuspend the cells by pipetting up and down with a 1-mL pipette.
-
ii. After the cells are resuspended, add 9 mL of 0.02% BSA/DNase.
-
-
36. Make a Nitex filter cone (see Movie 5), and pre-wet the filter with 1 mL of 0.02% BSA/DNase. Use flamed forceps (spray with ethanol and use a Bunsen flame to sterilize) to hold the Nitex filter in place.
-
37. Filter the resuspended cells through the Nitex mesh to eliminate remaining clumps of cells and chunks of tissue.
-
i. Filter 1 mL of resuspended cells at a time.
-
ii. After the cells have passed through the filter, wash it with 3 mL of 0.02% BSA/DNase.
-
-
38. Count the cells.
-
i. Dilute the cells 1:5 by combining 20 µL of cells with 80 µL of 0.02% BSA/DNase.
-
ii. Dilute this mixture 1:2 with trypan blue and count the cells using a hemacytometer.
-
There should be approximately 8–10 million cells per P7 pup at this stage.
-
To just isolate eGFP+ astrocytes from Aldh1L1-eGFP mice by FACS, eliminate the panning steps that follow and send the dissociated single-cell suspensions directly for FACS. Otherwise, continue to panning in Step 39.
-
-
Trituration. Illustrates Steps 27–32.
Filtering cells through nitex mesh. Illustrates Steps 36–41.
Panning
-
All panning steps should be done on a flat surface at room temperature.
-
39. Wash the first panning dish (the “secondary antibody only” dish) three times with DPBS (see Movie 5).
-
40. Add the filtered cells to secondary antibody panning dish. Swirl the dish to distribute the cells evenly. Let the dish sit for 7.5 min, gently swirl, and then leave to rest for another 7.5 min (15 min total).
-
Cells will adhere very quickly to this plate. Neuronal morphology is a good indicator of overall health; dissociated cells from P6–P8 mice should yield suspensions in which it is common to find neurons with small-to-medium length axons and dendrites.
-
-
41. Transfer the unbound cells (the supernatant) to the first “O4” dish. Wash the “secondary antibody only” dish once with 2 mL of 0.02% BSA/DNase, and add this buffer to the “O4” dish. Swirl the dish to distribute the cells evenly. Let the dish sit for 12.5 min, gently swirl, and then leave to rest for another 12.5 min (25 min total).
-
42. Transfer the unbound cells to the “PDGFRα” dish. Wash the O4 dish with 2 mL of 0.02% BSA/DNase, and add this buffer to the PDGFRα dish. Swirl the dish to distribute the cells evenly. Let the dish sit for 15 min, gently swirl, and then leave to rest for another 15 min (30 min total).
-
43. Transfer the unbound cells to the second “O4” dish. Do not wash the PDGFRα dish, because the cells only weakly adhere to this dish. Swirl the dish to distribute the cells evenly. Let the dish sit for 10 min, gently swirl, and then leave to rest for another 10 min (20 min total).
-
44. Transfer the unbound cells to a 50-mL conical tube.
-
45. Count the cells.
-
i. Dilute the cells 1:5 by combining 20 µL of cells with 80 µL of 0.02% BSA/DNase.
-
ii. Dilute this mixture 1:2 with trypan blue and count the cells using a hemacytometer.
-
For animals P8 and younger, proceed to Step 54 (FACS). For animals older than P8, that have begun to myelinate, perform additional depletion of oligodendrocytes and myelin in Steps 46–53 before continuing to FACS.
-
-
Additional Depletion for Animals Older than P8
-
Perform Steps 46–53 for additional depletion of oligodendrocytes and myelin.
-
46. Harvest the nonadherent cells from the second O4 dish by centrifugation at 170g for 5 min in a tabletop centrifuge at room temperature.
-
47. Resuspend the cells at 20 × 106 cells/5 mL (4 × 106 cells/mL) in 0.02% BSA/DNase containing 1:30 dilutions of galactocerebroside (GalC), myelin oligodendrocyte glycoprotein (MOG), and O1 hybridoma supernatants.
-
48. Incubate the cell suspension for 15 min at room temperature.
-
49. Wash the cells.
-
i. Dilute with 40 mL of 0.02% BSA/DNase.
-
ii. Centrifuge at 170g for 5 min in a tabletop centrifuge at room temperature. Aspirate and discard the supernatant.
-
-
50. Resuspend in 4 mL of 0.02% BSA/DNase containing 20 µg of donkey anti-mouse APC.
-
51. Incubate for 15 min at room temperature.
-
52. Wash out the secondary antibody by diluting the cells in 40 mL of 0.02% BSA/DNase.
-
53. Count the cells.
-
i. Dilute the cells 1:5 by combining 20 µL of cells with 80 µL of 0.02% BSA/DNase.
-
ii. Dilute this mixture 1:2 with trypan blue and count the cells using a hemacytometer.
-
Continue to Step 54 (FACS).
-
-
FACS
-
54. Centrifuge the cells at 170g for 5 min in a tabletop centrifuge at room temperature. Aspirate and discard the supernatant.
-
55. Resuspend the cells to a concentration of 10 × 106 cells/mL in 0.02% BSA/DNase with 8 µL of 1 mg/mL propidium iodide per 3 mL of cells, and sort the cells by FACS, as described by Cahoy et al. (2008).
-
56. Sort the cells using FACS, as described by Cahoy et al. (2008).
-
Each P7 pup brain should yield approximately 15 × 106 cells and ∼300,000 astrocytes.
-
Plating the Cells
-
57. After FACS, centrifuge the cells at 170g for 10 min in a tabletop centrifuge at room temperature.
-
58. Resuspend the cells in 0.02% BSA/DNase.
-
59. Plate the cells.
-
i. Carefully add 500 µl of IP-astrocyte base medium with HB-EGF to each well of a 24-well plate containing PDL-coated ACLAR plastic coverslips.
-
Alternatively, PDL-coated tissue culture dishes can be used without coverslips.
-
-
ii. Plate the cells at 10,000 cells/well.
-
iii. Centrifuge the plated cells at 40g for 3 min in a tabletop centrifuge at room temperature.
-
iv. Incubate the cells at 37°C in a 10% CO2 incubator.
-
ACKNOWLEDGMENTS
We thank Dr. Jim Huettner for helpful comments on the adaptation of his neuronal dissociation procedure to allow for glial purification.
- © 2013 Cold Spring Harbor Laboratory Press








