
An Optimized Protocol for High-Throughput In Situ Hybridization of Zebra Finch Brain
- Julia B. Carleton,
- Peter V. Lovell,
- Anne McHugh,
- Tessa Marzulla,
- Katy L. Horback and
- Claudio V. Mello1
Abstract
In situ hybridization (ISH) is a sensitive technique for documenting the tissue distribution of mRNAs. Advanced nonradioactive ISH methods that are based on the use of digoxigenin (DIG)-labeled probes and chromogenic detection have better spatial resolution than emulsion autoradiography techniques and, when paired with high-resolution digital imaging, allow for large-scale profiling of gene expression at cellular resolution within a histological context. However, technical challenges restrict the number of genes that can be investigated in a small laboratory setting. This protocol describes an optimized, low-cost, small-footprint, high-throughput ISH procedure to detect gene expression patterns in 10-µm brain sections from zebra finches. It uses DIG-labeled riboprobes synthesized from cDNA templates available through the Songbird Neurogenomics Consortium. The method is compatible with high-resolution digital imaging; it produces images with low background and a resolution approaching that of immunohistochemical methods. Approximately 180 slides can be processed each week using this protocol, but it can be scaled to accommodate a broad range of tissues from which cryosections can be obtained.
MATERIALS
Reagents
Acetylation buffer, freshly prepared
Agarose gel (1%; optional; see Step 7)
Anti-digoxigenin-AP antibody, Fab fragments (Roche 11093274910)
Aqueous mounting media (VectaMount AQ; Vector Labs H-5501)
-
We recommend VectaMount AQ. For most other brands, signal fades with time.
BCIP/NBT (chromogenic substrate for alkaline phosphatase; PerkinElmer NEL937)
BssHII restriction enzyme with buffer
Chloroform
Column blocking buffer, freshly prepared
DEPC-treated H2O
Dry ice
Ethanol series (70%, 95%, 100%)
Fixation buffer, freshly prepared
Heavy mineral (paraffin) oil
-
A staining container full of paraffin oil must be preequilibrated to 65°C in a water bath before use in Step 21.
Hybridization buffer for ISH, freshly prepared
Isopropanol
PCR purification kit (e.g., GeneJET PCR Purification Kit from Thermoscientific)
-
We recommend maintaining a dedicated kit that is nuclease-free.
Plasmid purification kit (e.g., GeneJET Plasmid Miniprep Kit from Thermoscientific)
RNA gel, prepared with DEPC-treated H2O (optional; see Step 12)
-
Dilute SSPE to 2× with DEPC-treated H2O before use.
T3 RNA polymerase (17 U/µL)
-
Commercially available plasmid vectors for in vitro transcription usually include a pair of polymerase binding sites that flank the cDNA insert: One is for sense strand RNA synthesis, and one is for antisense strand synthesis. Depending on the specific vector, these binding sites will either be for T3, T7, or SP6 polymerase. The majority of the clones we work with contain T7 (sense) and T3 (antisense) sites. However, users who require SP6 polymerase due to vector construction may find that a higher concentration of SP6 enzyme (i.e., >1.7 U/µL final concentration) is needed to produce RNA yields similar to that generated by T3 and/or T7. This can be accomplished by purchasing a high-concentration version of SP6.
Tissue-Tek O.C.T. compound (Sakura 4583)
-
Tissue-Tek provides optimal cutting and minimal freezing artifacts compared to other embedding media.
tRNA (Type X; 20 µg/µL in DEPC-treated H2O; Invitrogen 15401-011)
-
We use Type X tRNA; Types V and X-SA can contribute to high background.
Zebra finch cDNA templates for cDNA of interest and for control genes
-
This protocol was designed for templates from the Song Neurogenomics Consortium collection (http://titan.biotec.uiuc.edu/songbird/; Replogle et al. 2008), which can be purchased from the Clemson University Genomics Institute. In this collection, inserts were cloned into the pBluescriptII-SK+ vector, and can be released with flanking T3 and T7 promoters by BssHII digestion. However, inserts cloned in any vector containing RNA polymerase promoters can be used. Alternatively, templates can be generated by RT-PCR and then secondarily amplified using gene-specific primers with T7 (5'-GCGTAATACGACTCACTATAGGGCGAA-3’) and T3 (3'- GCGCAATTAACCCTCACTAAAGGGAAC-5’) overhangs. For culture and maintenance of bacterial stocks, cDNA cloning, and PCR amplification, see Sambrook et al. (1989).
Equipment
Agarose gel running apparatus (optional; see Step 7)
Centrifuge (minimum 1000 rcf; must fit 15-mL conical tubes; refrigeration optional)
Coverslip (22 × 40 mm)
Cryostat
Dissecting microscope
Glass wool (Sigma-Aldrich 18421), autoclaved
Humidified chamber
-
This can be purchased from commercial vendors, or constructed by placing wet paper towels into a 100 slide ABS plastic box (Fisherbrand 03-448-1). Each chamber can hold 14–16 slides resting side-up on the slotted guides.
Kimwipes
Microcentrifuge (must fit 1.5-mL microcentrifuge tubes; >12,000 rcf)
Microcentrifuge tubes (1.5-mL; nuclease-free preferred)
Orbital shaker (or equivalent)
PAP pen (e.g., Invitrogen 00-8877)
Paper towels
Parafilm (optional; see Step 4)
Razor blades
RNA gel apparatus (optional; see Step 12)
Round-bottom tubes (14-mL)
Serological pipettes
Slide boxes (for storage and humidity chambers)
Slide jars (Thermo Scientific 10 013 63)
Staining assemblies
-
Assemblies hold 20, 30, or 60 slides and consist of a stainless steel slide rack and container (Thermo Scientific). We recommend stainless steel, as opposed to glass or plastic, because it can be easily cleaned with water or solvents, is amenable to autoclaving, and is resistant to rapid changes in temperature.
Standard surgical dissection tools
Superfrost Plus slides
-
Charged slides minimize loss of sections during long high-temperature incubations.
Syringes (needle-free; 12-mL and 1-mL)
Syringe filters (0.22-µm; Millipore)
Thermometer
Tissue embedding molds (22 × 22 mm; Polysciences)
Water baths at 37°C, 50°C, and 65°C
METHOD
Sephadex G-50 Column Preparation
-
This procedure usually takes 30–60 min.
-
1. Roll autoclaved glass wool into small balls and place each ball into a 1-mL syringe barrel. Tamp down the glass wool with a syringe plunger.
-
2. Place each syringe into a 14-mL round-bottom tube. Use a serological pipette to add room temperature, TE-equilibrated, resuspended Sephadex G-50 into the syringe barrel. Centrifuge at 800 rcf for 60 sec at room temperature to pack the G-50 column. Discard the flowthrough. Repeat until the G-50 is ∼1 cm from the top of the syringe.
-
3. Add 200 µL of column blocking buffer and centrifuge for 2 min at 800 rcf. Discard the flowthrough.
-
4. Add 200 µL of column washing buffer and centrifuge for 2 min at 800 rcf. Discard the flowthrough. Repeat three times.
-
If desired, after this step add ∼150 µL column washing buffer to maintain hydration, Parafilm-wrap columns, and store at 4°C.
-
Riboprobe Preparation
-
This procedure usually takes 1 d.
-
5. Use a standard plasmid purification kit to purify plasmids containing cDNAs of interest, as well as plasmids containing control cDNAs.
-
As a positive control for in situ hybridization, we recommend including a plasmid with a cDNA previously shown to give a strong and reproducible signal. We routinely use GAD2, which yields high signal with low background.
-
-
6. Digest the plasmids with BssHII in the appropriate buffer for 3 h at 50°C to release the cDNA insert.
-
Aim to digest sufficient plasmid so that 0.6–0.8 µg of cDNA template will be available for use in Step 8.
-
-
7. Use a PCR purification kit to remove traces of enzymes and salts.
-
We usually recover the cDNA insert at a concentration of 0.15–0.2 µg/µL. We recommend running 1–2 µL on a 1% agarose gel to confirm digestion.
-
It is not necessary to separate the cDNA template from the plasmid vector. We use the DNA as is (i.e., at 0.15–0.2 µg/µL), rather than concentrating it.
-
-
8. Prepare the riboprobe synthesis buffer, and add 0.6–0.8 µg of cDNA template from Step 7, 1 µL of 17 U/µL T3 RNA polymerase, and DEPC-treated H2O to a final volume of 10 µL. Carry out the in vitro riboprobe synthesis reaction for 2–5 h at 37°C.
-
9. Remove a G-50 column from 4°C storage. Place the column into a 14-mL round-bottom tube and centrifuge at 800 rcf for 2 min at room temperature. Remove the flowthrough. Equilibrate the column by repeatedly adding 50 µL of column washing buffer, centrifuging at 800 rcf for 2 min, and discarding the flowthrough until a flowthrough volume of 50 µL is achieved, indicating that the column is equilibrated.
-
10. Dilute the reaction mix (10 µL) from Step 8 in 39 µL of column washing buffer and 1 µL of tRNA.
-
11. Remove the lid of a 1.5-mL microcentrifuge tube. Place the lid-less microcentrifuge tube into a 14-mL round-bottom tube. Place the equilibrated G-50 column into the lid-less microcentrifuge tube. Add the diluted in vitro reaction mix to the column. Centrifuge for 2 min at 800 rcf to purify the probe.
-
12. Remove the microcentrifuge tube from the round-bottom tube, and replace the lid. Store the probe at −80°C.
-
We do not routinely quantify probes, but recommend occasionally running an RNA gel to verify probe synthesis/integrity.
-
Tissue Preparation
-
This procedure usually takes 3–4 h.
-
13. Kill a bird via decapitation. Discard the dura and remove the brain tissue needed to produce the desired plane of section. Place the brain tissue into an embedding mold, cover with ice-cold Tissue-Tek, and freeze in a slurry of dry ice and isopropanol. Immediately proceed to Step 14 to section the tissue, or store the molds at −80°C.
-
Brains that take longer than 5 min to dissect and 2–3 min to freeze should be discarded, as tissue and mRNA quality may be compromised.
-
-
14. Cut 10-µm sections on a cryostat, and mount the sections on Superfrost Plus slides.
-
We recommend placing two adjacent sections on each slide for duplication.
-
-
15. Fill an appropriately sized staining assembly with freshly prepared fixation buffer, and fix the sections for 5 min at room temperature.
-
Longer fixation results in a marked decrease in signal.
-
-
16. Dehydrate the sections in an ascending ethanol series (70%, 95%, and 100% ethanol for 2 min each) in a staining assembly, air-dry the slides, and either proceed to hybridization (Step 17) or store long-term at −80°C.
Hybridization
-
This procedure usually takes 2–3 h, plus an overnight incubation.
-
17. Thaw the riboprobes from Step 12 on ice.
-
18. Acetylate the slides from Step 16 in a staining container containing freshly prepared acetylation buffer for 10 min at room temperature.
-
19. Rinse the slides twice in 2× SSPE, dehydrate in an ethanol series (70%, 95%, and 100% ethanol for 2 min each), and air-dry. Perform all of these procedures in a staining container.
-
20. Add 2 µL of riboprobe to a 30-µL volume of freshly prepared hybridization buffer, and pipette the entire 32-µL volume onto one slide. Place a single coverslip on the slide, covering both brain sections. Take care not to introduce air bubbles.
-
For each hybridization experiment, we recommend including one negative (no probe) and one positive (e.g., GAD2) control slide.
-
-
21. Load the slides horizontally into an 18-slide metal rack. Immerse the rack upright in the container of warm (65°C) paraffin oil.
-
22. Allow hybridization to proceed overnight (∼16 h).
Posthybridization Washes
-
This procedure usually takes ∼3 h.
-
23. Remove the slide rack from the oil, drain, and remove excess oil with paper towels.
-
24. Transfer the rack to chloroform at room temperature. Perform three 2-min incubations with fresh chloroform at room temperature to remove the remaining oil.
-
25. Remove the slides, evaporate the chloroform, and then immerse the rack in 2× SSPE in a staining container at room temperature. Gently jiggle each slide until the coverslip falls off. Transfer the slides to a new staining assembly filled with fresh 2× SSPE at room temperature.
-
Removing coverslips with forceps can damage tissue. Once the coverslips have been removed, the slides should be continuously immersed in aqueous buffer.
-
-
26. Place the rack in preheated (65°C) wash buffer I for 70 min. Agitate periodically (every 10 min).
-
For consistent results we place oil and wash buffer in the same water bath.
-
-
27. Transfer the slide rack to wash buffer II (65°C) and incubate for 30 min with periodic agitation. Repeat this step once with fresh wash buffer II.
-
28. Transfer the slide rack to a staining container with room-temperature TNT. Proceed to immunohistochemical detection or alternatively store overnight at 4°C.
Immunohistochemical Detection
-
This procedure usually takes 1–3 d.
-
29. For each slide, remove TNT from the glass area around the brain sections with a Kimwipe, and draw a waterproof border with a PAP pen.
-
Work quickly to ensure sections remain moist with TNT.
-
-
30. Pipette 200 µL of room-temperature TNB onto the sections.
-
31. Incubate the slide in a humidified chamber for 30 min at room temperature.
-
32. Discard the TNB by tapping the slide on a paper towel, and pipette 200 µL of TNB containing anti-digoxigenin-AP antibody (1:600) onto the slide. Incubate for 2 h at room temperature in a humidified chamber.
-
Titrate every new antibody batch, as nonoptimal dilutions can result in excessive background.
-
-
33. Discard the antibody solution, and wash the slide in TNM twice for 15 min at room temperature with agitation (on an orbital shaker or equivalent device).
-
34. Filter the BCIP/NBT solution into slide jars (30 mL per jar) with a 0.22-µm filter.
-
Filtering prevents deposition of artifacts on sections. If filter turns purple (chromogen oxidation), open fresh bottle of chromogen (BCIP/NBT).
-
-
35. Place the slides in jars, and incubate at room temperature with agitation.
-
Sealed/lightproof jars minimize chromogen oxidation, permitting longer incubations that facilitate the detection of low-abundance mRNAs.
-
-
36. Monitor the slides each day and stop the reaction when the signal is strong and the nonspecific labeling is minimal, usually after 1–3 d (see Fig. 1A–C).
-
Monitoring with a low-power compound (10× ocular, 4×–10× objective) or dissecting scope should be done quickly so as to avoid drying the sections. The negative control (no probe) slide should be completely devoid of signal. For genes with known expression patterns (e.g., GAD2), areas with no neuronal somata (e.g., fiber tracts) or no mature cells (e.g., ventricular region) should be devoid of signal (compare background and ventricular labeling in Figs. 1E1 and 1E2). See Troubleshooting.
-
-
37. Wash slides in deionized water for 1 h at room temperature with agitation.
-
38. Fix slides in fixation buffer for 10 min at room temperature, and then wash them twice for 15 min each time in deionized water at room temperature with agitation.
-
Fixing after probe detection prevents signal loss during long-term storage.
-
-
39. Scrape off the PAP pen border from each slide with a razor blade.
-
40. Air-dry the slides, add ∼400 µL of VectaMount AQ, and place a coverslip over the sections. Cure the slides 1–3 d before handling.
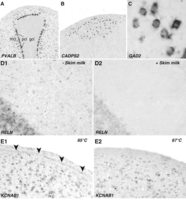
Sample results using an improved high-throughput ISH protocol for thin brain sections from zebra finches. (A–C) Examples of high-signal, low-background hybridizations, demonstrating the specificity and cellularity of neuronal labeling in zebra finch cerebellum (A), song nucleus HVC (B), and the medial striatum (C). (D) Compared to control sections reacted without skim milk (D1), the addition of 1% skim milk to the TNB blocking solution greatly reduces nonspecific background labeling, particularly with long (>3 d) chromogen incubations (D2). (E) Nonspecific labeling over the ventricle (E1, arrowheads) can be reduced by raising the temperature of the hybridization and posthybridization washes from 65°C to 67°C (E2). Abbreviations: gcl, granule cell layer; mcl, molecular cell label; pcl, Purkinje cell layer.
TROUBLESHOOTING
Problem (Step 36): The signal is strong, but nonspecific background is high.
Solution: The hybridization temperature is likely too low. Repeat the hybridization at a higher temperature (e.g., 67°C or 69°C) (see Fig. 1E), or increase the wash stringency by decreasing the SSPE concentration in wash buffer II to 0.01×. Alternatively, introduce an RNase treatment (30 min at 37°C; 1:1000 in 2× SSC) after posthybridization washes to further clean the pattern.
Problem (Step 36): No signal is detected following a 3-d incubation in chromogen.
Solution: The hybridization temperature is likely too high. Repeat the hybridization at a lower temperature (e.g., 63°C). For cross-species hybridizations, even lower temperatures may be required.
Problem (Step 36): Negative control (no probe) slide has high background staining.
Solution: The concentration of anti-DIG-AP is suboptimal. Perform an antibody titration on negative control slides to find an optimal dilution. Alternatively, increase the concentration of skim milk in TNB (see Fig. 1D).
Problem (Step 36): The labeling is strong, but sections show brown staining over fiber tracts.
Solution: The probe is sticky. Add a preincubation step using hybridization solution without riboprobe. In most cases a 60-min incubation at room temperature will reduce nonspecific labeling for sticky probes. Alternatively, introduce a posthybridization RNase treatment.
- © 2014 Cold Spring Harbor Laboratory Press








