
Reconstitution of Mice with Modified Hematopoietic Stem Cells
- Koch Institute for Integrative Cancer Research, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139
Abstract
Stem cell transplantation is well established in humans for the treatment of hematopoietic disease, including hematopoietic malignancies. Similar direct transplant procedures can readily be performed in mice; these procedures can be paired with retroviral infection to introduce exogenous genes or to silence endogenous genes in a subset of cells in the murine hematopoietic system. The resulting mice are chimeric for cells bearing a specific alteration. This approach has the advantage of examining tumorigenesis on a largely wild-type background (if only a subset of cells are infected), a situation that more accurately parallels the human situation. Additionally, tumor development occurs within the appropriate native microenvironment. Here, we describe the isolation and retroviral infection of hematopoietic stem cells (HSCs), as well as the reconstitution and monitoring of tumor formation in lethally irradiated recipient mice. This protocol requires a source of long-term HSCs; these can include either stimulated adult bone marrow or fetal liver—the site of primitive hematopoiesis.
MATERIALS
Reagents
5-Fluorouracil (5-FU)
Mice
-
The use of a CD45.1 strain is preferable, but not essential, for the isolation of HSCs. CD45.1 is a congenic strain used in transplant studies because it carries a differential B-cell antigen (originally designated Ly5.1 and CD45.1) that allows one to differentiate between donor and recipient cells. The b allele of Ptprc is normally present in the BALB and C57BL inbred strains.
Stem cell supplement (SCS, 5×)
Virus-packaging cells (Phoenix ecotropic or amphotropic)
Equipment
Cell strainers (40 µm)
Cesium-137 γ-cell irradiator
Conical tubes (15 mL)
Culture dishes (six well)
Flow cytometer
Flow cytometry tubes (round bottom) with mesh filter tops
Forceps
Frosted glass slides
Lineage-depletion kit
Needles (26.5 gauge)
Petri dishes
Surgical scalpels
Syringe filters (0.45 µm)
Syringes (1 mL and 5 mL)
Tissue-culture centrifuge
Tissue-culture hood
Tissue-culture incubator
METHOD
-
Perform Steps 1–11 if using liver cells as the source of HSCs or Steps 12–16 if using bone marrow. An illustrated flowchart of the steps in this procedure is shown in Figure 1.
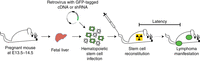
Tractable manipulation of the mouse hematopoietic system. Stem cell isolation, infection, and transplantation into immunodeficient recipient mice are shown. Both cDNAs and shRNAs can be introduced via retroviruses into HSCs. Additionally, diverse HSC genetic backgrounds can be used, allowing for the generation of combinatorially diverse chimeric models.
Isolation of HSCs from Fetal Liver
-
1. Set up timed matings of one male to two females per cage. Monitor female mice daily for vaginal plugs.
-
2. Kill pregnant female mice at E13.5–14.5.
-
Timing the liver harvest is critical, because HSCs migrate out of the fetal liver into the bone marrow starting at day 15.
-
-
3. Remove the entire uterine horn and place in B-cell medium in a Petri dish.
-
4. Transfer each embryo, still within its amniotic sac, into a separate Petri dish containing B-cell medium. Remove each embryo from its amniotic sac.
-
5. Remove the fetal liver from each embryo. Cut the embryo with a scalpel just above its liver, and squeeze the abdomen below the liver until the liver pushes out of the abdominal cavity into the B-cell medium.
-
The liver should be the only red-staining organ present.
-
-
6. Place each liver into a separate Petri dish with 5 mL of B-cell medium. Place a piece of the remaining embryo tissue into a microcentrifuge tube for genotyping.
-
7. Manually dissociate each fetal liver with frosted glass slides. Resuspend the cells and filter them through the lid of a blue fluorescence-activated cell sorter (FACS) tube cell strainer.
-
8. Centrifuge the fetal liver cells for 5 min in a 15-mL conical tube at 1500 rpm.
-
9. Resuspend each liver in 4 mL of 1× SCS and plate into four wells of a six-well dish.
-
Alternatively, at this point, fetal livers can be frozen and stored for future use. To freeze them, resuspend each fetal liver in 1 mL of B-cell freezing medium. Transfer to a labeled cryotube and freeze at −80°C. The next day, transfer to liquid nitrogen.
-
-
10. Culture fresh cells for 2 d at 37°C before infection. Proceed to Step 15.
-
Alternatively, to use frozen cells for infection, add cells to 10 mL of B-cell medium, invert the tube several times, and centrifuge for 5 min in a 15-mL conical tube at 1500 rpm. Resuspend each liver in 4 mL of 1× SCS and plate into four wells of a six-well dish. Culture the cells for 2 d at 37°C before infection.
-
Isolation of HSCs from Bone Marrow
-
11. Inject 4-wk-old mice with 150 mg/kg 5-FU to induce proliferation of hematopoietic stem cells.
-
12. Six d later, collect the long leg bones (femur and tibia) of the injected mice and crush with a sterile mortar and pestle in a tissue-culture hood.
-
13. Add 2 mL of B-cell medium per mouse and pipette up and down thoroughly to liberate cells from the associated bone. Filter the resuspended cells through a 40-µm cell strainer.
-
14. Centrifuge the cell suspension at 1500 rpm for 5 min. Resuspend the cell pellet in 1 mL of 1× SCS per mouse. Plate 1 mL/well in a six-well dish. Culture the cells for 1 d at 37°C before infection. Proceed to Step 15.
Infection
-
15. During the preparation of fetal liver or bone marrow populations, produce viruses in packaging cells such that peak viral production occurs during the infection period (Swift et al. 2001).
-
Use the same strategy for lentiviral and retroviral infections.
-
-
16. Collect viral supernatant from the packaging cells in B-cell medium. Add fresh B-cell medium and collect a second viral fraction later in the day.
-
17. Filter the collected viral supernatant through a 0.45-μm filter.
-
18. Add 5× SCS to the appropriate volume of viral supernatants to make 1× SCS. Add polybrene to a final concentration of 4 µg/mL.
-
For each infection, only 0.75 mL (0.6 mL virus plus 0.15 mL 5× SCS) will be added to each well of a six-well dish, so only make enough 1× SCS to perform infections.
-
-
19. Add 0.75 mL of the virus/well to the fetal stem cells and centrifuge at 1500 rpm for 5 min in the six-well dishes.
-
This will infect the cells with the virus.
-
-
20. Repeat Steps 16–19 three more times at 10–12 h intervals for a total of four infections over 2 d. After the final infection, culture the cells for 2 d at 37°C before injection.
-
See Troubleshooting.
-
Injection
-
21. Before injecting the infected cells into mice, irradiate the recipient mice with split dose 10 Gy whole-body irradiation (5 Gy + 5 Gy, split by 4 h) with a cesium-137 γ-cell radiation source.
-
Independent calculation of a lethal dose is necessary with the use of any γ-cell irradiator.
-
-
22. Resuspend the fetal liver/bone marrow cells in 0.5 mL PBS and determine the cell concentration. Inject one to two million stem and progenitor cells into the tail vein of each mouse. (This number of cells should be present in approximately one well of a six-well plate.)
-
The actual number of HSCs can be determined with greater accuracy using lineage exclusion columns and flow cytometry analysis using stem cell markers. Additionally, HSC populations can be enriched before injection, if purer populations are required. If pure stem and progenitor populations are injected, coinjection of a similar number of uninfected whole bone marrow cells may be necessary to provide some hematopoietic function before stem cell engraftment.
-
-
23. Measure the infection efficiency by flow cytometry.
-
It is helpful to use retroviral vectors containing fluorescent markers.
-
-
24. Monitor mouse reconstitution by examining the number of CD45.1+ or fluorescently labeled cells that are present in the circulating leukocyte population 8–10 wk after injection.
-
See Troubleshooting.
-
Troubleshooting
Problem (Step 20): There is poor infection.
Solution: Low infection efficiency can arise because of poor virus production or poor target cell growth. In cases of poor infection, viral stocks should be titered before infection. Additionally, cell growth should be monitored during HSC culture. Cells should appear healthy at the time of viral addition, and the overall cell number should expand considerably during cell culture.
Problem (Step 24): There is poor reconstitution (i.e., absence of CD45.1+ or fluorescently labeled cells in reconstituted mice) or the animal dies.
Solution: Poor reconstitution can occur if the recipient immune system is not adequately ablated. If long-term HSCs are not injected into recipient, high levels of recipient death will be observed within a month of reconstitution. Injected populations of cells should be examined by (1) quantitation after lineage depletion and (2) cell surface marker expression for the presence of stem and progenitor cells. Absence of stem cells can occur if the starting fetal livers are too old or if the SCS does not contain the appropriate cytokine mix.
DISCUSSION
Stem cell transduction followed by mouse adoptive transfer represents an effective strategy for assessing the role of single genes or combinations of genes in tumor progression (Pear et al. 1998; Schmitt et al. 2000). Paired with RNAi technology, this strategy can be used to examine both putative tumor suppressors and oncogenes. Thus, complex sets of alterations that recapitulate bona fide tumor genotypes can be rapidly generated. This approach does, however, have some notable limitations. First, with constitutive expression of a retroviral oncogene throughout the reconstituted hematopoietic system, it can be difficult to resolve when the gene actually contributes to tumor development (Gilbert and Hemann 2012). For example, a gene may promote stem cell survival or expansion, but not transformation of a more mature target cell. Second, retroviral vectors can promote tumor development via integration proximal to a proto-oncogene. Thus, injection of a large cohort of mice with control vector-infected cells is necessary to draw definitive conclusions regarding the candidacy of a putative oncogene.
ACKNOWLEDGMENTS
Much of the approach described here was adapted and modified from the work of Warren Pear and Clemens Schmitt.
- © 2015 Cold Spring Harbor Laboratory Press








