
Metabolomic Analysis of Schizosaccharomyces pombe: Sample Preparation, Detection, and Data Interpretation
- 1G0 Cell Unit, Okinawa Institute of Science and Technology Graduate University (OIST), Onna-son, Kunigami, Okinawa 904-0495, Japan
Abstract
Metabolomics is a modern field of chemical biology that strives to simultaneously quantify hundreds of cellular metabolites. Techniques for metabolomic analysis in Schizosaccharomyces pombe have only recently been developed. Here we introduce methods that provide a complete workflow for metabolomic analysis in S. pombe. Based on available literature, we estimate the yeast metabolome to comprise on the order of several thousand different metabolites. We discuss the feasibility of extraction and detection of such a large number of metabolites, and the influences of various parameters on the results. Among the parameters addressed are cell cultivation conditions, metabolite extraction techniques, and detection and quantification methods. Further, we provide recommendations on data management and data processing for metabolomic experiments, and describe possible pitfalls regarding the interpretation of metabolomic data. Finally, we briefly discuss potential future developments of this technique.
INTRODUCTION
Metabolites are small molecules (often defined as MW <1 kDa) involved in biochemical pathways. In conjunction with enzymes, they are integrated into broad, cell-wide metabolic networks. Cellular metabolites are divided into primary metabolites, which are compounds directly involved in normal growth, development, and reproduction, and secondary metabolites, which are not essential for survival and are often specific to particular species. Metabolomics aims to qualitatively and quantitatively profile all metabolites, collectively known as the metabolome (Patti et al. 2012). While individual reactions and pathways of cellular metabolism have been extensively studied by biologists for centuries, serious large-scale metabolomic analyses have only become possible in recent years following technical advances in measurement technologies. Among the levels in the hierarchy known as the central dogma of molecular biology (Fig. 1), the metabolome comes closest to the cellular phenotype, thus enabling detection of otherwise unnoticeable phenotypic changes, identification of biomarkers, and analysis of gene functions (Harrigan and Goodacre 2003). Apart from genetic control, basic cellular processes are regulated by the extracellular environment and nutrition. Understanding cells at the metabolic level is therefore vital to study cellular phenomena such as aging, and for modeling metabolic diseases such as diabetes.
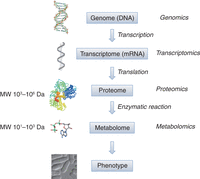
The central dogma of molecular biology showing the basic concept of information transfer from genome (DNA) to phenotype. Related omics research fields are shown on the right.
The first reference to S. pombe metabolomics occurred in a study of the MAP-kinase deletion mutant sty1-Δ under oxidative stress. The study used a combination of proteomic analysis and small-scale metabolomic analysis (29 metabolites) using 1H-NMR (Weeks et al. 2006). Later, we reported the identification and semiquantitative measurement of 123 metabolites using liquid chromatography–mass spectrometry (LC–MS) (Pluskal et al. 2010b). We found an accumulation of trehalose, glycerophosphoethanolamine, arabitol, ribulose, and ophthalmic acid under heat stress. We also confirmed the absence of ferrichrome in the deletion mutant of ferrichrome synthetase, Δsib1, and characterized extensive perturbations to the metabolome of an HMG-CoA synthase temperature-sensitive mutant, hcs1-143. There have been several additional studies. Takeda et al. (2010) measured the accumulation of the cellular antioxidants glutathione and ergothioneine in the proteasome regulatory subunit mutant mts3-1 in G0 state. We described the metabolic effects of glucose starvation and identified several specific metabolites (e.g., biotin, trehalose, ergothioneine, S-adenosyl-methionine and CDP-choline) as biomarkers for different glucose concentrations in the culture medium (Pluskal et al. 2011). Nakamura et al. (2012) measured intermediates of the coenzyme A synthesis pathway from pantothenate, and described the accumulation of 4′-phosphopantothenate in the phosphopantothenoylcysteine synthetase mutant ppc1-537. Shimanuki et al. (2013) reported accumulation of several metabolites, including N-acetyl-D-glucosaminate, ergothioneine, and S-adenosyl-methionine, in the deletion mutant of C2H2 zinc finger transcription factor Δklf1 under long-term quiescence. Sajiki et al. (2013) characterized swift transformation of the yeast metabolome during the first hour of nitrogen starvation, manifested mainly by rapid shutdown of purine metabolism, decreased free amino acids, and increased 2-oxoglutarate, trehalose, succinate, and hercynylcysteine sulfoxide. Finally, we combined the use of genetics and metabolomics to dissect the biosynthetic pathway of ergothioneine and selenoineine (Pluskal et al. 2014).
OVERVIEW OF THE YEAST METABOLOME
Unlike the genome, the information about metabolites is not encoded in a single, readable entity such as DNA. Because new chemical structures are produced from existing compounds by enzymes or by interactions with reactive molecules such as free radicals, we can only estimate the total number of distinct metabolites present in the cell. Furthermore, the metabolic profile of any given cell is highly dependent on the extracellular environment, especially the chemical composition and abundance of available nutrients.
Scale of the Yeast Metabolome
The Yeast Metabolome Database (YMDB) is an online curated resource which annotates all metabolites reported from the budding yeast (Jewison et al. 2012). This database currently contains 2027 metabolite entries. The METLIN metabolite database, which is not restricted to any one species, contains 242,766 (Smith et al. 2005). Alternative estimates of the extent of the yeast metabolome can be obtained from reconstructed genome-wide metabolic models. The latest version of the S. cerevisiae metabolic network model (v. 7.11; available at http://yeast.sf.net) contains 2386 compounds (Aung et al. 2013), while the S. pombe genome-scale metabolic model SpoMBEL1693 contains 1744 (Sohn et al. 2012). It should be noted, however, that because of the nature of these systems biology models, identical chemical compounds present in different cellular compartments (e.g., ATP in cytoplasm versus ATP in mitochondria) are counted as different metabolites. Contrarily, some entities in the models are described using “Markush” structures containing R-groups; therefore, they represent heterogeneous chemical compounds. In conclusion, the total number of distinct metabolites present in a yeast cell probably is on the order of several thousand.
The wide range of metabolite concentrations means that metabolomic studies rarely report more than ~150 of the thousands of different metabolites. The most abundant metabolites such as ATP or glutathione are present in mm-level concentrations (Ingram and Barnes 2000; Song and Lim 2008), while the least abundant are below 1 nm (assuming several molecules per intracellular volume of ~150 fL). This dynamic range of seven orders of magnitude presents a challenge for even the most sensitive detection methods. Furthermore, some metabolites are only synthesized under particular cellular conditions. Another considerable challenge is presented by a need to separate individual isomers (e.g., glucose, galactose, fructose, mannose, etc.) or highly similar compounds (e.g., leucine versus isoleucine). Nevertheless, an empirical observation shared by many researchers is that in contemporary metabolomic studies, a majority of detected signals still await identification.
Metabolic Pathway Databases
Knowledge about relationships among individual metabolites in the global metabolic network is essential for successful interpretation of metabolomic data. Various online databases collect and curate such information, such as the Kyoto Encyclopedia of Genes and Genomes (KEGG; Kanehisa and Goto 2000), MetaCyc (Caspi et al. 2014), and the Small Molecule Pathway Database (SMPDB; Jewison et al. 2014). KEGG was the first database to comprehensively cover relationships among genes, enzymes, and metabolites, and arguably represents the most complete and convenient available resource about both ubiquitous and species-specific metabolic pathways.
METABOLOMIC ANALYSIS
Metabolomic analysis typically follows an established workflow (Fig. 2). We provide two protocols which encompass the complete workflow for metabolomic analysis in fission yeast: Protocol: Preparation of Intracellular Metabolite Extracts from Liquid Schizosaccharomyces pombe Cultures (Pluskal et al. 2016) describes intracellular metabolite extraction from liquid cell cultures, optimized mainly for polar metabolites. Protocol: Measurement of Metabolome Samples Using Liquid Chromatography–Mass Spectrometry, Data Acquisition, and Processing (Pluskal and Yanagida 2016) describes semiquantitative measurement of metabolomic samples, including the steps required for data analysis.
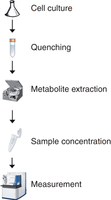
Typical workflow of a metabolomic experiment.
In brief, starting from a liquid cell culture, cells are rapidly quenched to provide an immediate snapshot of the metabolome. The subsequent sample preparation step is performed at low temperature to prevent further chemical reactions. Finally, prepared samples are analyzed, providing qualitative and quantitative data about extracted metabolites.
Metabolomic Sample Preparation
Cell Cultivation
For metabolomic analysis, it is highly advisable to cultivate cells in a well-defined synthetic culture medium, such as EMM2 (see Introduction: Growth and the Environment of Schizosaccharomyces pombe [Petersen and Russell 2016]). Rich and undefined media with unknown chemical composition, such as YES (based on yeast extract) or YPD (based on yeast extract and polypeptone), cannot guarantee reproducible metabolic conditions among experiments, much less among laboratories.
Cultivation temperature has a profound effect on the cellular metabolome (Pluskal et al. 2010b). Wild-type fission yeast can grow in the range of approximately 18°C–36°C, with cells at higher temperatures showing shorter division times. Generally, ~26°C is considered the least stressful for S. pombe cells, and is also optimal for growing temperature-sensitive mutants. Thus, it is recommended for metabolomic sampling unless there is an experimental reason to use a different temperature.
The number of cells required to prepare a metabolite extract depends on the sensitivity of the instruments used for detection. Approximately 2 × 108 cells (e.g., 40 mL of cell culture cultivated to mid-log phase, or 5 × 106 cells/mL) is a good starting point for an initial analysis. This number can be altered according to the results of pilot experiments. It is important to consider, however, that the total volume of intracellular fluid should be normalized between samples, making average cell size a key parameter to take into account. The average cell size depends on the strain, DNA content (haploid/diploid), culture medium, and cultivation conditions. For example, the average size of nitrogen-starved S. pombe G0 cells is about threefold smaller than that of vegetatively growing cells. To adjust for this difference, we commonly use 3× more nitrogen-starved cells than vegetative cells for metabolomic sampling. Alternatively, measurement of optical density (OD) at 595 nm can be used to standardize protein content (see Introduction: Growth and the Environment of Schizosaccharomyces pombe [Petersen and Russell 2016]).
Quenching
The unceasing activity of a metabolic network can be likened to the vigorous buzzing of a Middle Eastern bazaar. Various metabolites chaotically move through cellular compartments and interact with other molecules, while guided by enzymes and transporter proteins. Turnover rates of individual compounds can be very rapid. To obtain reliable and reproducible measurements of the metabolome, cell cultures must thus be instantly quenched. Note that in this regard, the metabolome differs from the genome, which does not change, and from transcriptomes and proteomes, which do change but on a much longer timescale.
The most commonly applied method for rapid quenching of the metabolome in budding yeast—plunging cells into a 60% methanol solution at −40°C (de Koning and van Dam 1992)—has been superseded by the use of 100% methanol to prevent metabolite leakage (Canelas et al. 2008). We recommend pure methanol at −40°C as a quenching solution for S. pombe.
Metabolite Extraction
For the extraction of intracellular metabolites from budding yeast, a number of protocols have been developed (Dunn and Winder 2011). Unfortunately, because of a huge variation in physical and chemical properties, as well as intracellular concentrations, no single method can guarantee an efficient extraction of all classes of metabolites. Villas-Bôas et al. (2005) compared six different methods for the extraction of intracellular metabolites and concluded that freeze–thawing in liquid nitrogen followed by centrifugation in pure methanol (−40°C) provided the best overall recovery of a wide range of metabolites. However, Canelas et al. (2009) reevaluated available extraction methods and found that freeze–thawing in methanol did not ensure proper inactivation of enzymatic activity. They therefore recommended the use of boiling ethanol or chloroform–methanol extractions. Sporty et al. (2008) developed a protocol for measurement of NAD and NADH in which cell lysis was achieved by bead-beating in ice-cold ammonium acetate. Recently, Kim et al. (2013) proposed acetonitrile/water (1:1 v/v) at −20°C as a more efficient extraction solvent.
As there is no optimal protocol to extract and measure all metabolites, we shall hereafter focus on the analysis of polar metabolites, such as amino acids, organic acids, nucleotides, sugars, and sugar-phosphates. It is important to stress that very different approaches must be used to measure nonpolar metabolites, such as lipids or fatty acids. For example, shotgun mass spectrometry (MS) has been applied to study lipids in S. cerevisiae (Ejsing et al. 2009; Klose et al. 2012).
Our metabolite extraction protocol for S. pombe (see Protocol: Preparation of Intracellular Metabolite Extracts from Liquid Schizosaccharomyces pombe Cultures [Pluskal et al. 2016]) was developed with the goal that it be easily applicable to a wide range of cell culture volumes (i.e., from a few milliliters for quick screening experiments to several liters for the purpose of purification of individual metabolites). We therefore adopted a combination of fast filtration with cold methanol quenching. First, cell cultures are vacuum-filtered on a methanol-resistant membrane filter, and the filter with the cell biomass is then dropped into 25 mL of pure methanol at −40°C.
After quenching, internal standards should be added to each sample to facilitate the verification of metabolite extraction. At the data processing stage, normalization of all signals by the signal intensity of the internal standards reduces the effect of daily instrument variation. A low concentration of stable exogenous compounds such as buffers can be used as internal standards. We commonly add 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and piperazine-N,N′-bis(2-ethanesulfonic acid (PIPES), each at 10 nmol.
A metabolite extraction protocol must allow an efficient extraction of metabolites from any cellular condition. Fission yeast cells can develop stress resistance, including the reorganization of the cell wall, following exposure to heat, oxidative stress, or starvation. We found that metabolites from vegetatively grown or glucose-starved S. pombe cells could be efficiently extracted by centrifugation in cold 50% methanol, as showed with propidium iodide (PI) staining (Fig. 3A,C). However, PI staining of nitrogen-starved quiescent cells was considerably less intense, indicating inefficient disruption of cellular membranes (Fig. 3B). We therefore recommend using a bead-beating system, such as Multi-beads Shocker (Yasui Kikai) or Geno/Grinder (SPEX SamplePrep), to disrupt cell walls and membranes and efficiently extract intracellular metabolites, regardless of cell condition.
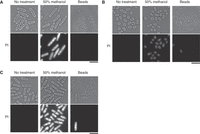
Micrographs of vegetative (A), nitrogen-starved (B) and glucose-starved (C) fission yeast cells stained with propidium iodide (PI), which binds to nucleic acids. Cells were cultivated for 24 h in an appropriate medium (EMM2, EMM2-N, or EMM2 with 1.1 mm glucose, respectively) and centrifuged in cold 50% methanol (center column), optionally followed by bead-beating (right column). Bright PI staining indicates leakage of nucleic acids, and successful disruption of cellular membranes. Bar = 10 µm.
Following the extraction of metabolites, proteins and other crude cellular components must be removed from the sample before analysis. This can be achieved thoroughly by filtration with a 10 kDa cutoff filter. (Smaller cutoffs such as 3 kDa are also available, but result in longer filtration times.) As a final step, filtered samples can be concentrated by vacuum evaporation and resuspended in a solvent suitable for detection. This step improves detection of less abundant metabolites and also ensures a constant final sample volume, essential for reproducible quantitative measurements. For MS, 50% acetonitrile is an efficient final solvent.
Metabolite Detection and Quantification
Metabolite Detection
The easiest, albeit costly, way to measure the metabolite levels in extracted samples is to outsource measurement to a commercial source such as Metabolon Inc., Human Metabolome Technologies, or Metabolomic Discoveries GmbH. These companies typically return absolute (molar) concentrations of up to hundreds of characterized metabolites, alongside a semiquantitative comparison of unknown signals and their structural analysis.
Two technologies are commonly applied for in-house measurement of metabolomic samples: nuclear magnetic resonance (NMR) and MS, the latter typically in combination with gas or liquid chromatography (GC–MS or LC–MS) or capillary electrophoresis (CE–MS) (Dunn et al. 2011). The advantages of NMR are simple sample preparation, rapid and nondestructive analysis, and the ability to obtain structural information about purified molecules. On the other hand, identification and quantification of less abundant metabolites in complex NMR spectra is challenging, making this technology optimal for quick fingerprinting of samples. For microbial metabolomics, MS is a more convenient detection technology because it provides superior sensitivity by isolating mass profiles of individual metabolites for quantification and structural analysis (via tandem MS, or MS/MS). Recently, high-resolution MS detectors, which provide mass accuracy in the range of several ppm, have become standard. Such high mass accuracy, combined with measurement of isotopic distributions, typically allows the determination of molecular formulas of the majority of measured compounds (Pluskal et al. 2012). Older MS instruments with unit mass accuracy are not suitable for metabolomics.
Good chromatographic separation is essential to achieve reliable quantification of metabolites. Diverse chemical properties and high polarity of cellular metabolites must be taken into account. For example, measuring adenosine triphosphate (ATP) by GC–MS is inherently difficult (as it is difficult to derivatize it to a volatile form), while its measurement by reverse phase LC–MS typically requires buffers incompatible with MS analysis. We present a liquid chromatography detection protocol based on hydrophilic interaction chromatography (HILIC) combined with measurement using a high-resolution Orbitrap MS detector (see Protocol: Measurement of Metabolome Samples Using Liquid Chromatography-Mass Spectrometry, Data Acquisition, and Processing [Pluskal and Yanagida 2016] and example data in Fig. 4A). This protocol is suitable for simultaneous measurement and identification of hundreds of polar metabolites (including ATP), and provides good long-term stability and reproducibility. (We have used it for over 7 yr.) The benefits of HILIC separation combined with MS detection have been reviewed in detail by Nguyen and Schug (2008). It should be noted, though, that complementary approaches using different LC–MS (Zhou et al. 2012), GC–MS (Garcia and Barbas 2011), or CE–MS (Ramautar et al. 2009) methods are available, and the decision to apply a particular technology should be based on its applicability to target molecules and on the instrumentation available to the researcher, as well as the researcher’s expertise in its use.
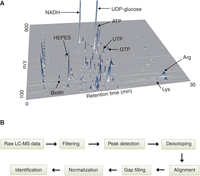
(A) Example of an LC–MS data set acquired using described methods. Several prominent metabolites are annotated. (B) Typical workflow for processing metabolomics LC–MS data. Optional steps are indicated by dashed lines.
One aspect of LC–MS metabolite detection that remains challenging is the separation of isomeric compounds, such as various hexose sugars, which cannot be distinguished by MS alone. While the above-mentioned HILIC-based protocol can separate the peaks of various sugar isomers in a single experiment, their retention times are very similar and in practice it becomes difficult to align corresponding signals from multiple data sets. If the experimental goal requires reliable quantification of such isomeric compounds, a specialized chromatography method should be applied.
Data Management and Processing
For large-scale metabolomic studies, the matter of data storage and archiving must be carefully considered. A single high-resolution LC–MS or GC–MS run can produce raw data totaling tens to hundreds of megabytes. Because each sample is often analyzed multiple times (e.g., using different ionization modes or different chromatography methods), the data produced from a single sample can easily exceed a gigabyte. A study containing 1000 samples requires tens of terabytes of data storage to accommodate the sample analysis runs, control and test runs, pure standard runs, data analyses, and backups.
Raw LC–MS data must first be translated into lists of metabolites with one of a number of software tools (Castillo et al. 2011; Sugimoto et al. 2012). Our data processing protocol employs the free MZmine 2 framework for MS data visualization and analysis (Pluskal et al. 2010a) (see Protocol: Measurement of Metabolome Samples Using Liquid Chromatography-Mass Spectrometry, Data Acquisition, and Processing [Pluskal and Yanagida 2016] and Fig. 4B). Briefly, depending on the MS instrument, raw data may first be filtered for smoothing or baseline correction. Then a peak detection algorithm is applied, producing a list of peaks found in each file, characterized by their retention times, m/z values, and areas (for quantification). Each peak list is filtered for isotopic signals produced by naturally occurring isotopes of common elements (such as 13C, 15N, 34S, etc.). These isotopic peaks are removed from the peak lists, although information about the intensity of each isotope is retained because it provides a valuable clue for chemical formula identification. Peak lists generated from each raw data file are then aligned to match corresponding signals with the same m/z ratios and retention times. In the aligned data sets, gaps (misaligned or undetected peaks) may be filled by secondary peak detection, and intensity signals are normalized using internal standards. Finally, metabolites are identified using their mass (or m/z), retention times, and fragmentation (MS/MS) spectra, if available. Pure standards are usually applied to verify the authenticity of reported compounds. If MS/MS spectra are available, comparison with spectral databases such as MassBank (Horai et al. 2010) and METLIN (Smith et al. 2005) can also greatly assist with the verification of compound identity. An integrated identification strategy called MetFusion was recently developed (Gerlich and Neumann 2013), combining spectral database searches with in silico fragmentation prediction.
For publication of metabolomic experiments, minimal reporting standards have been developed and should be followed accordingly (Goodacre et al. 2007; van der Werf et al. 2007). Acquired MS raw data can be stored in public repositories such as MetabolomeExpress (Carroll et al. 2010) or MetaboLights (Haug et al. 2013).
Data Interpretation
As MS measurements can provide quantitative data on thousands of features in a large number of samples, the discovery of significant features that differentiate certain conditions requires sophisticated statistical analysis (Brown et al. 2005). Univariate and multivariate techniques are commonly applied to metabolome data sets (Liland 2011; Vinaixa et al. 2012). An online service called MetaboAnalyst implements the common methods of statistical analysis and quality control, as well as metabolic pathway visualization (Xia et al. 2012).
Biological interpretation of metabolomic data is often not straightforward and underlying mechanisms must therefore be carefully dissected. An increase or decrease in the concentration of a particular metabolite might not necessarily mean the activity of the major pathway involving this metabolite is up- or down-regulated. A change in metabolic flux can often cause secondary effects, perturbing the whole metabolic network. On the other hand, when interpreted with care, the high-throughput nature of metabolome data can provide extremely useful insights into the conditions inside cells.
SUMMARY
Metabolomics is a frontier technique that still suffers from a lack of standardization and immature software tools; however, technology is improving rapidly and many exciting research projects are on the way. The two protocols we provide have been thoroughly validated in several published studies.
Several directions are available for the future development of this field. One aspect that has not been deeply covered in microorganisms (Paczia et al. 2012)—and remains virtually untouched in fission yeast—is that of the extracellular metabolome (exometabolome), including excretion of secondary metabolites (Krug and Muller 2014). Another interesting topic is the analysis of metabolic fluxes, also called fluxomics (Mo et al. 2009; Celton et al. 2012). In budding yeast, the first single-cell metabolomic studies have recently appeared (Ibanez et al. 2013; Zenobi 2013), and methods for high-throughput screening of low-volume cultures from 96-well plates are also being developed (Ewald et al. 2009).
ACKNOWLEDGMENTS
We thank Dr. Steven D. Aird for editing the manuscript. We acknowledge the generous funding and support of Okinawa Institute of Science and Technology Promotion Corporation and Okinawa Institute of Science and Technology Graduate University.
Footnotes
-
↵3 Correspondence: pluskal{at}wi.mit.edu
-
From the Fission Yeast collection, edited by Iain M. Hagan, Antony M. Carr, Agnes Grallert, and Paul Nurse.
- © 2016 Cold Spring Harbor Laboratory Press








