
Genome Editing in Human Pluripotent Stem Cells
- Cory Smith1,2,3,
- Zhaohui Ye1,2 and
- Linzhao Cheng1,2,3,4
- 1Division of Hematology, Department of Medicine, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205;
- 2Stem Cell Program, Institute for Cell Engineering, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205;
- 3Predoctoral Training Program in Human Genetics, Johns Hopkins University School of Medicine, Baltimore, Maryland 21205
Abstract
Pluripotent stem cells (PSCs), defined by their capacity for self-renewal and differentiation into all cell types, are an integral tool for basic biological research and disease modeling. However, full use of PSCs for research and regenerative medicine requires the ability to precisely edit their DNA to correct disease-causing mutations and for functional analysis of genetic variations. Recent advances in DNA editing of human stem cells (including PSCs) have benefited from the use of designer nucleases capable of making double-strand breaks (DSBs) at specific sequences that stimulate endogenous DNA repair. The clustered, regularly interspaced short palindromic repeats (CRISPR)–Cas9 system has become the preferred designer nuclease for genome editing in human PSCs and other cell types. Here we describe the principles for designing a single guide RNA to uniquely target a gene of interest and describe strategies for disrupting, inserting, or replacing a specific DNA sequence in human PSCs. The improvements in efficiency and ease provided by these techniques allow individuals to precisely engineer PSCs in a way previously limited to large institutes and core facilities.
EMBRYONIC AND PLURIPOTENT STEM CELLS
Embryonic stem cells (ESCs) are derived from the inner cell mass of a developing blastocyst and can be expanded in vitro for decades without losing their full potential for differentiation and self-renewal. ESCs hold great potential for basic biological research, disease modeling, and as a cell source for regenerative medicine. However, because of the destructive nature of their derivation, an autologous source of ESCs is not available without therapeutic cloning. Also, the high monetary and ethical costs associated with obtaining oocytes limit the practical applications and scalability of their use. The discovery that four simple transcription factors could reprogram somatic fibroblasts to an embryonic-like state (i.e., induced PSCs) provides an autologous cell source for individualized regenerative medicine and basic research, including disease modeling, high-throughput drug screening, and access to human cell types previously unavailable for live study, including neurons and astrocytes. The full realization of these PSCs will rely on the ability to precisely manipulate their DNA to investigate the effects of these variants in isolation or to cure a disease-causing variant ex vivo before cell-based regenerative therapy.
Gene targeting using a double-stranded DNA donor with long homology arms (1–4 kb) is a well-established technique across many animal models and cell lines. Although this approach has been used in mouse PSCs, the many technical limitations using human PSCs prevented successful modification of this technique: Single cells rarely survive without cell-to-cell contacts and signals that aid in the formation of the early colony after the stress of electroporation, and the rates of homologous recombination are significantly lower in human PSCs than in mice. The first limitation was addressed by the development of Rho-associated kinase inhibitor that dramatically increases survival and proper colony formation after the single-cell digestion of human PSCs required for electroporation; the second issue was addressed by the development of sequence-specific nucleases that generate a DSB in DNA at a targeted location, increasing the efficiency of recombination at that site by several orders of magnitude (Hockemeyer et al. 2009; Zou et al. 2009). DSBs are detected by the cell’s endogenous DNA repair machinery, which proceeds through either the error-prone nonhomologous end joining (NHEJ) pathway that often results in small indel mutations around the break site or by homology-dependent repair (HDR) using a DNA donor template. These tools allow researchers to both disrupt genes through NHEJ and to create single-nucleotide variants with an HDR donor to determine its effects in isolation when compared with an otherwise isogenic cell line.
CRISPR–Cas EDITING OF PSCs
The Streptococcus pyogenes–derived adaptive bacterial immune system CRISPR–Cas degrades foreign DNA in a sequence-specific manner guided by short guide RNAs (gRNAs) to induce a DSB. In particular, the type II CRISPR system only requires a single protein component, Cas9, and a single gRNA to determine its target and cleave DNA. This system has been successfully adapted for mammalian expression (Cong et al. 2013; Mali et al. 2013). A major advantage of CRISPR–Cas9 is the ability to rapidly design, synthesize, assemble, and test gRNAs targeting new genes or noncoding regions of interest, as well as the ability to multiplex by transfecting additional gRNAs. Several studies—largely conducted using cancer cell lines (e.g., HEK 293T or U20S)—revealed higher than expected off-target mutagenesis, including some sites that were altered more frequently than the intended target and other sites that included up to five mismatches compared with the gRNA (Fu et al. 2013; Hsu et al. 2013; Mali et al. 2013); further screening in animal embryos and human stem cells reported minimal off-target effects (Wang et al. 2013; Niu et al. 2014; Wu et al. 2014). Several complementary approaches to screen human PSCs for potential off-target mutagenesis such as targeted sequencing of top sites predicted most likely to cleave based on sequence similarity revealed high specificity of these gRNAs tested in human PSCs relative to HEK 293Ts (Mali et al. 2013; Smith et al. 2014; Li et al. 2015). Additionally, whole-genome sequencing conducted on CRISPR–Cas9-targeted PSCs revealed new mutations in each clone, but none were similar to the gRNA target site or recurrent between the clones, indicating that genome editing of PSCs can be performed with minimal off-target mutagenesis genome-wide (Smith et al. 2014; Suzuki et al. 2014; Veres et al. 2014; Yang et al. 2014).
There are many approaches for genome editing to either knock out a gene, induce a specific single-nucleotide change, or insert a larger fragment for transgene expression or to add a fluorescent tag (Fig. 1). The most basic strategy involves a single guide RNA targeting the protein coding sequence of a gene to disrupt expression by a frameshift mutation, often leading to degradation through nonsense-mediated decay. If two gRNAs are used in close proximity (~20 bp–10 kb) the intervening sequences is deleted, often with precise breakpoints 3 nucleotides upstream of the NGG, although small indels are observed around the junction at a low frequency. This technique can be used to delete exons that could not otherwise be targeted because of highly conserved domains and also provides a tool to excise defined regions, making it a promising approach to investigate noncoding variants such as regulatory elements over a broad range of deletion sizes. To induce specific mutations (e.g., single-nucleotide changes), the classic approach uses a double-stranded DNA donor plasmid with homology arms of ~0.5–2 kb. Using CRISPR–Cas9, this approach can produce dramatically higher efficiencies (~10%) without using drug selection (Byrne et al. 2015) and can also be used to insert transgenes into a predetermined safe harbor locus (e.g., the adeno-associated virus integration site 1) or to tag a lineage-specific gene to monitor expression during differentiation. Another targeting approach uses a single-stranded DNA oligonucleotide (ssODN) of ~70–110 bases, although 90 bp was optimal in human PSCs (Yang et al. 2013). The same study also reported that it was ideal to use the strand complementary to the gRNA for the ssODN as opposed to the same strand. It is best to design the gRNA as close to the mutation as possible (ideally, within 10 nucleotides for ssODNs or 200 nucleotides for plasmid donors) (Xie et al. 2014).
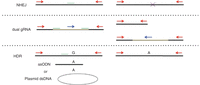
Targeting approaches for genome editing. NHEJ uses a single guide RNA (green) targeting the protein coding sequence of a gene to disrupt expression by a frameshift mutation (×). Dual guide RNA uses two gRNAs in close proximity to delete the intervening sequences (blue arrow). HDR can be used with either a single-stranded DNA oligonucleotide (ssODN) or a double-stranded DNA donor (plasmid dsDNA) to induce specific mutations.
In Protocol: Protocol for Genome Editing in Human Pluripotent Stem Cells (Smith et al. 2016), we describe a method for genome editing in PSCs with CRISPR–Cas9 to disrupt or delete a predetermined DNA sequence or to alter a nucleotide of interest for further study. Targeting considerations are discussed to design gRNAs for both knockout and knock-in strategies that have the least chance of off-target mutagenesis predicted bioinformatically and tested experimentally in the PSC line of interest. We also provide a simplified procedure for gRNA synthesis and assembly and discuss considerations for transfection-quality DNA needed to modify the traditionally difficult-to-transfect PSCs. Briefly, transfection is achieved through electroporation using the Nucleofector 4D (Lonza), although similar efficiencies in PSCs can be obtained using other technologies (e.g., Life Technologies’ Neon Transfection System). With initially ~1% NHEJ disruption efficiencies for Cas9 in human PSCs (Mali et al. 2013) clonal isolation and screening for targeted clones remains the most time-consuming and costly part of the procedure as hundreds to thousands of clones need to be screened to find several with the intended mutation. Although feasible, several improvements have been made to increase the efficiency of editing by using either fluorescence-assisted cell sorting for Cas9_GFP+ cells (Ding et al. 2013) or by integrating a dox-inducible Cas9 system (González et al. 2014). The future application of these genome-editing techniques to PSCs will allow cell-based functional investigations of any genetic variant of interest such as the incoming torrent of genome-wide association data or potential causative variants in rare case anomalies at the extremes of phenotypes.
- © 2016 Cold Spring Harbor Laboratory Press








