
The Use of Lysosomotropic Dyes to Exclude Lysosomal Membrane Permeabilization
- 1Department of Biochemistry and Molecular and Structural Biology, J. Stefan Institute, SI-1000 Ljubljana, Slovenia;
- 2Department of Biosciences, University of Oslo, NO-0371 Oslo, Norway;
- 3Center of Excellence CIPKEBIP, SI-1000 Ljubljana, Slovenia
Abstract
Progressive lowering of pH is characteristic for the endocytic pathway and enables efficient degradation of molecules by hydrolytic enzymes at its distal end. The existence of the proton gradient over the endosomal/lysosomal membranes depends on the action of the vacuolar ATPase (v-ATPase). During lysosomal membrane permeabilization (LMP), protons leak through the destabilized membrane, resulting in loss of the pH gradient. Here, we present a protocol showing how this effect can be detected by staining cells with lysosomotropic dyes, which accumulate in acidic organelles after protonation. During LMP, cells lose the ability to retain these dyes and therefore appear pale. Among the most commonly used lysosomotropic dyes are LysoTracker reagents and acridine orange. Cells can be analyzed with a fluorescence microscope; however, flow-cytometric analysis enables fast, objective, and reliable evaluation of differences between samples. Advantages of the technique include the fact that sample preparation is relatively simple and can be scaled-up to test several different compounds or conditions. However, as we will discuss, cells treated with v-ATPase inhibitors also lose the pH gradient across lysosomal membranes and cannot be stained with lysosomotropic dyes, although this is not accompanied by LMP. Therefore, merely observing loss of staining is not in itself a proof of LMP.
MATERIALS
Reagents
Acridine orange (1 mg/mL stock in distilled water)
Bafilomycin A1 (optional; see Step 2)
Cell-detachment reagent (e.g., TrypLE Select, Invitrogen/Life Technologies)
Cell source
-
Stocks (e.g., HeLa cells) to be used both as an experimental sample and as a reference sample (untreated cells).
Leu-Leu-O-methylester (LLOMe) (optional; see Step 2)
LysoTracker reagents (1 mm stocks in DMSO)
-
LysoTracker Green DND-26 (Molecular Probes/Life Technologies, L7526)
-
LysoTracker Red DND-99 (Molecular Probes/Life Technologies, L7528)
-
See Discussion.
-
Phosphate-buffered saline (PBS[D])
Equipment
Centrifuge
CO2 incubator
Flow cytometer (with a blue 488 nm laser, such as FACSCalibur, BD Biosciences)
Fluorescence-activated cell sorter (FACS) tubes (3.5 mL, for flow-cytometric analysis)
-
Note that the volumes given in the text below are relevant for the FACSCalibur flow cytometer (BD Biosciences).
Fluorescence microscope (wide-field or confocal)
Glass-bottom cell-culture dishes (for light-microscopic analysis)
Pipette
Plates (24-well)
METHOD
Preparation of Cells
-
The following protocol is for adherent cells; note that, as the cells can detach upon treatment, the supernatant containing detached cells should also be collected during sample preparation. However, the protocol works equally well for cells in suspension—this is simpler as no cell detachment is needed.
-
1. Seed cells in a 24-well plate and allow to attach overnight at their normal growth temperature in a CO2 incubator.
-
The number of cells seeded and the volume of the cell culture supernatant should correspond to the growth area of the dish relative to other types of dishes used. With practice, two 24-plates can be prepared in parallel.
-
-
2. Perform the treatment necessary for the experimental aims. Also run a positive control for reduced staining: For example, apply bafilomycin A1 (30 nm) for a minimum of 30 min to inhibit the vacuolar ATPase (v-ATPase) and/or the lysosomal membrane permeabilization (LMP)-inducing compound LLOMe (1–5 mm) for 15 min.
-
3. Treat the cells in the 24-well plate with a lysosomotropic dye by adding it to the cell culture medium and incubating for the appropriate period of time at 37°C (e.g., for mammalian cells, use 40 nm LysoTracker Green for 5–10 min or 1 µg/mL acridine orange for 15 min).
-
4. After incubation with the dye, analyze cells with a fluorescence microscope or with a flow cytometer (see the next steps). For microscopic analysis, wash cells 3× with warm PBS and add imaging medium. For flow cytometry analysis, begin to prepare a cell suspension. To this end, transfer the cell culture supernatant into a 3.5-mL FACS tube, wash the well with 0.5 mL of warm (at room temperature) PBS, and add the wash to the supernatant in the FACS tube.
-
5. Add a small volume (sufficient just to cover the cells) of a cell-detachment reagent (e.g., TrypLE Select) to the wells with attached cells and incubate at 37°C untill the cells detach.
-
6. Transfer the detached cells to the FACS tube containing the culture supernatant from Step 4.
-
7. Centrifuge the cells (e.g., at 300g for 5 min at room temperature). With a pipette, carefully remove and discard the supernatant.
-
8. Resuspend the pellet in 300–500 µL of warm (room temperature) PBS. Analyze immediately with a flow cytometer.
FACS Analysis
-
9. Select settings for forward and side scatter (FSC, SSC) so that the cell population in the FSC/SSC plot is central and can be delineated from the debris and dead cells. Set a gate around the population of viable cells.
-
10. Open a histogram plot to measure fluorescence. For LysoTracker Green, select the green channel (530/30 bandpass filter), and, for acridine orange, select the far-red channel (>670 nm).
-
11. Set the voltage on the detectors so that the main peak of stained untreated control cells lies between values of 102 and 103, which enables the detection of increased and decreased fluorescence in treated samples.
-
If cells are stained only with the lysosomotropic dye, no compensation to correct for the overlap between emission spectra of different fluorochromes is required.
-
-
12. Collect between 5000 and 10,000 events (cells), and analyze the data (see Fig. 1).
-
Set a gate around live cells in a FSC/SSC plot (Fig. 1A) to exclude debris and dead cells, which have a reduced FSC signal. Fluorescence data can be presented as frequency histograms of fluorescence intensity (Fig. 1, B and D). For semiquantitative analysis the geometric mean rather than simple mean values can be read, especially when the histograms are asymmetric. The geometric mean data can be statistically analyzed and presented in a bar plot (Fig. 1, C and E). To allow a direct comparison of geometric mean values, samples must be stained in parallel and analyzed with the same settings. Results can also be presented relative to untreated control cells (as a fraction of the fluorescence intensity of control cells). When cells in the population behave in more than one way, the percentage of pale cells can be determined and the geometric mean value of this subpopulation can be read.
-
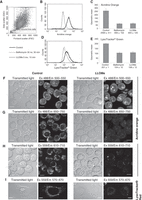
Lysosomal membrane permeabilization (LMP) causes the loss of the pH gradient over lysosomal membranes, and as a result cells show reduced staining with lysosomotropic dyes compared with untreated control cells. (A–E) HeLa cells, untreated or cells treated with 30 nm bafilomycin A1 (v-ATPase inhibitor) for 30 min, or with 5 mm Leu-Leu-O-methylester (LLOMe; a known LMP inducer) for 15 min, were stained with acridine orange or LysoTracker Green and analyzed with a flow cytometer. Before the fluorescence analysis, the gate was set around live cells in a density plot of forward and side scatter (FSC/SSC) to exclude debris. Results of the acridine orange or Lysotracker Green staining are presented as frequency histograms of fluorescence intensity (B,C) and as mean ±s.d. values for the geometric mean of fluorescence intensity (D,E). The experiment was performed in duplicate. (F–I) RAW264.7 cells, stained with acridine orange (F–H), and HeLa cells, stained with Lysotracker Red (I), were analyzed with a confocal laser scanning microscope. Cells stained with acridine orange were analyzed with three different combinations of excitation and emission light. Paired untreated control and LLOMe-treated samples were analyzed at the same imaging settings. Scale bar, 10 µm.
DISCUSSION
Choice of Dye
Various LysoTracker reagents with different excitation and emission spectra are available. LysoTracker Green is suitable for flow-cytometric analysis because it absorbs at 488 nm, and most of the flow cytometry machines are equipped with an appropriate blue laser. For fluorescence microscopy, in our experience, LysoTracker Red is more convenient because it is more stable, whereas LysoTracker Green bleaches quickly and LysoTracker Blue is rather faint. Alternatively, acridine orange dye can be used, which is particularly suitable for the analysis with a flow cytometer because the blue laser and the far-red detector are compatible with the analysis of its metachromatic emission. This is characteristic of protonated acridine orange molecules in acidic organelles, which form aggregates whose emission maximum changes to far-red, compared to orthochromatic green emission maximum of the monomeric dye (Darzynkiewicz and Kapuscinski 1990). The terms orthochromatic and metachromatic imply that spectral properties of the dye on binding to the target do not change and do change, respectively.
In our experience, prolonged incubation with LysoTracker dyes can result in nonselective staining of cellular structures other than lysosomes. We incubate for 15 min with LysoTracker Red and acridine orange, and only 5–10 min with LysoTracker Green. Even if stained cells are analyzed with flow cytometry, stained samples should be inspected in the fluorescence microscope during the protocol optimization.
Lysosomes of dead cells are not stained, thus, especially in samples taken late after the experimental treatment, cell viability should also be analyzed to discriminate live cells, with pale lysosomes, from dead cells. Dead cells can be distinguished from viable cells on the basis of altered light-scatter properties; they generally show a decrease in forward-scatter and an increase in side-scatter signals (Loken and Herzenberg 1975). If this is the case, they should be excluded from the analysis gate in a FSC/SSC plot. An increased fraction of dead cells in the sample can indicate damage to organelles other than lysosomes, and this should be investigated to understand the role of LMP in cell death signaling.
LMP can be transient, in which case the lysosomal membrane regains its integrity and rapidly restores the acidic pH (Steinberg et al. 2010). Transient LMP can therefore be showed during a limited period of time. Finally, note that if experimentally tested compounds are fluorescent and their emission spectra overlap with those of lysosomotropic dyes, the cells treated with them cannot be analyzed with this protocol.
Experimental Tactics and Caveats
Cells with decreased fluorescence in comparison with untreated controls, also termed “pale cells,” represent cells with lost or reduced ability to retain acidic dyes. This can be the consequence of either LMP or the inhibition of the v-ATPase—and use of this method alone does not allow the two causes to be discriminated. In contrast, an increased fluorescent signal indicates an increase in the volume of the endosomal compartments that occurs owing to increased number or size of the acidic organelles. For example, chloroquine and NH4Cl increase the volume of the endosomes owing to the so-called proton-sponge effect.
In our experience, acridine orange gives a stronger signal and a stronger lysosomal signal:background ratio compared with LysoTracker Green (Fig. 1, B and D), which makes it a more sensitive dye. Acridine orange is a metachromatic dye and, as such, emits two colors. When excited with blue light, monomeric acridine orange emits orthochromatic green. When acridine orange accumulates in lysosomes at a high concentration, it precipitates into oligomeric structures that show a shift in the emission to metachromatic red at 640 nm (Darzynkiewicz and Kapuscinski 1990).
To detect LMP, it is possible to stain cells with acridine orange before the treatment and monitor the loss of red fluorescence and the concomitant increase in green fluorescence when the dye is released into the cytosol (Huai et al. 2013). However, there are several limitations to this approach. First, in the case of incubations lasting several hours, acridine orange that is loaded in lysosomes will interfere with their function. Second, the increase in green fluorescence is considerably smaller than the decrease in red fluorescence (Huai et al. 2013), which lowers the sensitivity of the assay. Third, this method is not compatible with lysosomotropic compounds, whose accumulation in the lysosomal lumen depends on protonation and thereby low pH. Acridine orange, like LysoTracker, is a lysosomotropic compound and its accumulation in lysosomes raises pH, which is likely to interfere with the accumulation of a lysosomotropic LMP-inducing compound, such as LLOMe.
An Experimental Example
To show the versatility of acridine orange for acidic organelle staining, we have labeled untreated control RAW264.7 cells or cells that were treated with the LMP inducer LLOMe (5 mm) with acridine orange for 15 min and analyzed them with a confocal laser scanning microscope. When excited with a 488 nm laser, acidic organelles in control cells emit green light. Nuclei also emit green, and there is considerable green background in the cytosol. In cells with destabilized lysosomal membranes, the punctate signal in the cytoplasm is no longer present, whereas nuclei and the cytoplasm are stained (Fig. 1F). If lysosomes were stained with acridine orange before LMP was induced, the dye released into the cytosol should increase the green cytosolic signal. However, this increase is relatively small and difficult to show given the strong background signal. The metachromatic emission is shown in Fig. 1G. When excited with blue light, the far-red signal represents acidic organelles. This detection corresponds to the detection analyzed with a flow cytometer (Fig. 1, B and C). Acidic organelles stained with acridine orange can also be observed with green excitation—for example, with a 559 nm laser. The emitted light is weaker compared with the excitation at 488 nm, but the signal is also predominantly localized to the acidic organelles (Fig. 1H). After LMP, the punctate staining is lost. Similarly, untreated control HeLa cells stained with Lysotracker Red show a punctate pattern representing acidic organelles, whereas cells that have experienced LMP appear pale (Fig. 1I).
Concluding Remarks
In summary, although staining with lysosomotropic dyes is a rather simple method, it is nevertheless extremely powerful. Sample preparation is straightforward and flow-cytometric analysis is fast, objective, statistically strong, and allows high throughput. This all makes this protocol a good starting point to screen different compounds or different experimental conditions and perhaps the simplest approach to determine the exact time-point when LMP occurs.
ACKNOWLEDGMENTS
We thank the NorMIC Imaging Platform at the Department of Biosciences, University of Oslo, and especially Cathrine Heyward for discussion and assistance with the imaging.
Footnotes
-
↵4 Correspondence: urska.repnik{at}ibv.uio.no; boris.turk{at}ijs.si
- © 2016 Cold Spring Harbor Laboratory Press








