
Detecting Single-Nucleotide Substitutions Induced by Genome Editing
- 1Gladstone Institute of Cardiovascular Disease, San Francisco, California 94158;
- 2Departments of Medicine, and Cellular and Molecular Pharmacology, University of California, San Francisco, California 94143
Abstract
The detection of genome editing is critical in evaluating genome-editing tools or conditions, but it is not an easy task to detect genome-editing events—especially single-nucleotide substitutions—without a surrogate marker. Here we introduce a procedure that significantly contributes to the advancement of genome-editing technologies. It uses droplet digital polymerase chain reaction (ddPCR) and allele-specific hydrolysis probes to detect single-nucleotide substitutions generated by genome editing (via homology-directed repair, or HDR). HDR events that introduce substitutions using donor DNA are generally infrequent, even with genome-editing tools, and the outcome is only one base pair difference in 3 billion base pairs of the human genome. This task is particularly difficult in induced pluripotent stem (iPS) cells, in which editing events can be very rare. Therefore, the technological advances described here have implications for therapeutic genome editing and experimental approaches to disease modeling with iPS cells.
THE CHALLENGE OF DETECTING HOMOLOGY-DIRECTED REPAIR EVENTS IN iPS CELLS
The discovery of human induced pluripotent stem (iPS) cells (Takahashi et al. 2007; Yu et al. 2007) provided a great opportunity to study human disease phenotypes in vitro. However, iPS-cell disease modeling was limited to observational studies, because iPS cells were resistant to the homologous recombination methods used for mouse embryonic stem cells. Site-specific nucleases (Gaj et al. 2013) have revolutionized our ability to make isogenic iPS-cell models that exactly reflect patients’ pathological mutations, or to precisely revert pathological mutations to a healthy sequence. Many human diseases are caused by a single point mutation, so it is critical to make accurate disease models with as few additional changes as possible. Traditional gene targeting methods to generate mutant cell lines use antibiotic-resistance markers, leaving a genetic “scar” that can interfere with studying the resulting phenotype (da Cunha Santos et al. 2011; Moore et al. 2012). A major challenge of nuclease-driven precise mutagenesis in iPS cells is that the mutations can be <1% of the cells (Chen et al. 2011; Soldner et al. 2011; Ding et al. 2013), and isolating the right recombinant iPS clones is difficult without antibiotic selection. Furthermore, using high levels of nuclease or increasing nuclease activity runs the risk of reducing the fidelity of mutagenesis (Gupta et al. 2011; Pattanayak et al. 2011; Fu et al. 2013; Hsu et al. 2013).
As multiple groups are now planning to use genome editing to correct point mutations for therapeutic purposes, it becomes even more critical to develop methods that accurately measure homology-directed repair (HDR) events with a wide dynamic range. Initial studies (Chen et al. 2011; Soldner et al. 2011; Ding et al. 2013) suggest that the highest-fidelity editing conditions would favor using lower concentrations of nucleases or replacing them with nickases to avoid off-target mutations (Mali et al. 2013; Ran et al. 2013). However, using nickases to improve fidelity also lowers efficiency so that mutants are increasingly rare. Therefore, genome engineering is faced with a logistical challenge: Precise mutagenesis with the highest fidelity results in rarer mutagenic events, but isolating a rare mutant cell without antibiotic resistance amid the hundreds of otherwise identical cells is exceedingly difficult.
DROPLET DIGITAL POLYMERASE CHAIN REACTION FOR DETECTING HDR EVENTS
In the accompanying protocol, we provide a method that uses droplet digital polymerase chain reaction (ddPCR) (Hindson et al. 2011) to efficiently and quantitatively detect rare single-base mutations for precise genome editing (see Protocol: Using Digital Polymerase Chain Reaction to Detect Single-Nucleotide Substitutions Induced by Genome Editing [Miyaoka et al. 2016]). ddPCR detects, in a robust manner, single-nucleotide substitutions that are introduced by HDR, even in cell types that are difficult to engineer, such as iPS cells. It partitions a reaction into more than 10,000 nanoliter-scale water-in-oil droplets, each of which contains only a few copies of the genome. In this way, ddPCR detects rare genome-editing events.
Because of its high sensitivity and quantitative performance, this method can be used to validate genome-editing tools, isolate cell lines with single-nucleotide substitutions, and perform genotyping (Miyaoka et al. 2014). Substitution of single nucleotides in human cells, especially human pluripotent stem cells, is a highly valuable approach for disease modeling and future cell therapies. This method will empower scientists to fully exploit the advantages of genome-editing tools.
THE FUTURE OF GENOME EDITING IN iPS CELLS
Perhaps the most immediate impact of genome editing in iPS cells is in disease modeling, because it provides a clear path to experiments with isogenic controls. The value of isogenic controls is well known to researchers who work in model organisms, such as mice, in which inbreed strains are considered essential for well-controlled experiments. Human pluripotent stem cells were so difficult to engineer that only eight gene-targeting events were reported within 10 years after the first isolation of human embryonic stem cells (Giudice and Trounson 2008). After the discovery of human iPS cells (Takahashi et al. 2007; Yu et al. 2007), the difficulties with genome editing limited disease modeling in iPS cells to observations without isogenic controls. The advent of transcription-activator-like effector nucleases (TALENs), the CRISPR–Cas9 system, and the methods we describe here has dramatically changed disease modeling with iPS cells (Fig. 1A).
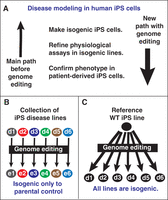
Examples of how genome editing has changed the landscape for phenotyping human iPS cells. (A) Before genome-editing methods became robust, scientists made iPS-cell disease lines and attempted to develop in vitro assays with imperfect nonisogenic controls (up arrow). Prior to 2009, making isogenic controls was very difficult; it was only done in a few cases. Now that genome-editing methods are robust (larger down arrow), scientists can start with isogenic controls, making assay development much easier. Once the assays are developed, they can be used on the more challenging patient-derived iPS-cell lines. (B) Illustration of how collections of patient-derived iPS-cell disease lines derived from patients (d1–d6) can be genome-engineered (e1–e6), providing valuable controls. However, these collections of engineered iPS cells have different genetic backgrounds (indicated by the different colors), so they do not provide an isogenic allelic series. (C) Illustration of how using genome engineering on a common reference line results in an allelic series with a common genetic background. Although the number of engineered lines is the same in B and C, engineering a common reference line (C) is much easier, because a single robust iPS line, rather than multiple patient lines (each with potentially different culture conditions), can be chosen. In addition, there is scientific benefit in having an isogenic allelic series. WT, wild type.
Our own initial studies with iPS cells from patients with a disease, long QT syndrome (Spencer et al. 2014), made us aware that using nonisogenic controls impeded our efforts to develop assays that bring out the disease phenotype (Fig. 1A). We found that each patient iPS-cell line can have unique physiological features that are driven by their genetic background, making it harder to determine the physiological effects that were solely due to the disease mutation. With the advent of genome editing, we can make putative disease mutations in a reference cell line so that assay development can proceed in an isogenic background. Once the assays have been optimized, it is much easier to examine the more challenging patient-derived lines (Fig. 1A). In our experience, making the isogenic iPS lines from a robust reference cell line dramatically decreases the time and cost of a study.
Precise genome editing in iPS cells also has ramifications for how the community approaches large-scale collections of iPS cells harboring genetic diseases. In the past, the only route to making these iPS-cell collections was to collect large numbers of patient samples (Fig. 1B). In fact, multiple international efforts have been launched to build large collections of diverse iPS-cell lines, with more than 100,000 lines currently planned for cell banks around the world (Soares et al. 2014). These collections will certainly be of value in the future. However, it is now possible to consider parallel collections of iPS cells that are made from common reference cell lines (Fig. 1C). The advantage of this approach is that each disease line is isogenic with other disease lines and provides an allele series. Furthermore, because multiple gene-editing events can be achieved, gene–gene interactions can be examined to allow for complex traits to be observed on a controlled, isogenic background. This approach is similar to the large collections of gene-targeted mice that have been made on common genetic backgrounds. Although the reference banks of patient-derived iPS cells will have value, the rapid advance of precise genome-editing methods will provide a complementary approach for each investigator who is embarking on a study with iPS cells.
In summary, we introduced a method that will empower scientists to make and detect scarless single-base mutations in many cell types, including human iPS cells. This method provides a wealth of new opportunities to exploit advantages of genome-editing tools and use iPS cells for disease modeling.
- © 2016 Cold Spring Harbor Laboratory Press








