
Methods to Synthesize Large DNA Fragments for a Synthetic Yeast Genome
- 1Daniel Rutherford Building G.24, School of Biological Sciences, University of Edinburgh, The King’s Buildings, Edinburgh EH9 3BF, United Kingdom;
- 2MOE Key Laboratory of Bioinformatics, Center for Synthetic and Systems Biology, School of Life Sciences, Tsinghua University, Beijing 100084, People’s Republic of China
- ↵3Correspondence: yizhi.cai{at}ed.ac.uk; jbdai{at}tsinghua.edu.cn
Abstract
De novo DNA synthesis is one of the key enabling technologies for synthetic biology. Methods for large-scale DNA synthesis, in particular, have transformed many facets of life science research, supporting new discoveries in biology through the design of novel synthetic biological systems. This protocol describes in detail the methods currently being used to synthesize and assemble large pieces of DNA for the synthetic yeast genome project. The protocol includes instructions for building block synthesis as well as chunk assembly, each of which can be used as a stand-alone procedure to generate a synthetic DNA of interest.
MATERIALS
Reagents
Escherichia coli cells, chemically competent
Low-salt LB (Lysogeny Broth) liquid medium
-
Prepare low-salt LB liquid medium with kanamycin for Step 19 by adding 1 mL of filtered kanamycin stock solution (40 mg/mL in ddH2O; 1000×) per liter of medium.
Reagents for building block synthesis (Steps 1–24)
-
Agarose gel (1%) and reagents for gel electrophoresis
-
GoTaq Green Master Mix (Promega)
-
High-fidelity polymerase chain reaction (PCR) amplification kit (e.g., Phusion High-Fidelity PCR Kit; New England Biolabs)
-
M13 primers
-
M13 forward (M13F; 5′-GTAAAACGACGGCCAG-3′)
-
M13 reverse (M13R; 5′-CAGGAAACAGCTATGAC-3′)
-
-
Overlapping oligonucleotides for building block synthesis (300 nm)
-
Overlapping oligonucleotides of a target synthetic DNA sequence can be designed using the Building Block design module (constant length overlap) of the software GeneDesign (www.genedesign.org) (Richardson et al. 2006, 2010, 2012). A 20-bp overlap is typically used; however, the overlap length can be optimized for different target sequences. Oligonucleotides (normally 40–80 bp in length) can be synthesized in-house or purchased from commercial vendors and must be diluted to a concentration of 300 nm. To avoid the extra work and error-prone procedures of primer dilution, we recommend ordering primers as prediluted liquids from the vendor.
-
In our initial design, we incorporated building blocks of ∼750 bp in length. The purpose of limiting the length of the target sequences was to ensure that one round of Sanger sequencing was adequate to cover the entire sequence, and building blocks were sequenced from both ends to ensure there were no mutations on either strand. If single-strand sequencing is sufficient, building blocks can be as long as 1600–2000 bp. Building block length can also be adjusted to meet various needs, such as the incorporation of restriction enzyme recognition sites.
-
-
Zero Blunt TOPO PCR Cloning Kit, with pCR Blunt II-TOPO cloning vector and salt solution (1.2 m NaCl, 0.06 m MgCl2) (Invitrogen)
Reagents for chunk assembly (Steps 25–30)
-
Backbone vector (e.g., pRS415), linearized by restriction digestion
-
To minimize background from the undigested backbone, we recommend gel purification of the vector digestion before assembly.
-
-
Building blocks for assembly
-
Each building block should contain at least 40 bp of terminal overlap with its adjacent building blocks. The first building block should contain a 40-bp 5′ overlap with the linearized vector backbone. Building block inserts can be amplified via PCR or digested from the plasmids constructed in the first section of this protocol (“Synthesizing Building Blocks from Oligonucleotides” [Steps 1–24]).
-
-
Isothermal reaction (Gibson Assembly) master mix
-
This protocol uses homemade isothermal reaction master mix for Gibson Assembly; however, a similar product can be purchased from commercial vendors.
-
-
Plates containing selective solid growth medium
-
Choose a selective medium according to the vector backbone. For pRS vectors, use plates containing LB medium with carbenicillin.
-
-
Yeast selectable marker (such as Leu2 or Ura3)
-
PCR-amplify the yeast selectable marker such that it contains a 40-bp 5′ terminal overlap with the last building block and a 40-bp 3′ terminal overlap with the linearized vector backbone.
-
Equipment
96-well plates (sterile, with lids)
Gel electrophoresis equipment
Glass beads
Heat block or water bath at 42°C
Incubators at 30°C and 37°C
Microcentrifuge tubes
PCR tubes
Thermal cycler
METHOD
Synthesizing Building Blocks from Oligonucleotides
-
The protocol described here is suitable for high-throughput processing and is ready for automation.
-
If you are using methods other than GeneDesign for oligonucleotide design, minor adjustments to the PCR programs according to your design parameters may be required for high-quality results.
Preparing Overlapping Oligonucleotides
-
1. Add 10 µL of each overlapping oligonucleotide to a microcentrifuge tube to prepare a templateless primer mix (TPM). If there are fewer than 20 oligos, add H2O to bring the final volume to 200 µL. Mix well.
-
2. Add 25 µL of the first and last oligonucleotides in the sequence to a microcentrifuge tube to prepare an outer oligo mix (OPM). Mix well.
Performing Templateless PCR (TPCR)
-
3. For each TPCR, assemble the following reaction using a high-fidelity PCR amplification kit. Mix well and keep on ice.
Reagent Volume dNTPs (2.5 mm) 2.0 µL 10× reaction buffer 2.5 µL High-fidelity DNA polymerase 0.25 µL H2O 17.75 µL TPM (∼300 nm) 2.5 µL Total volume 25 µL -
4. Perform TPCR using the following program.
Number of cycles Time Temperature 1 3 min 94°C 30 sec 55°C 1 min 72°C 5 30 sec 94°C 30 sec 69°C 1 min 72°C 5 30 sec 94°C 30 sec 65°C 1 min 72°C 20 30 sec 94°C 30 sec 61°C 1 min/kb 72°C 1 3 min 72°C Hold 4°C
Performing Finish PCR (FPCR)
-
5. Dilute each TPCR sample 1:5 in H2O.
-
6. For each FPCR, assemble the following reaction using a high-fidelity PCR amplification kit. Mix well and keep on ice.
Reagent Volume dNTPs (2.5 mm) 2.0 µL 10× reaction buffer 2.5 µL High-fidelity DNA polymerase 0.25 µL H2O 15.75 µL Diluted (1:5) TPM product 2.5 µL OPM (∼300 nm) 2.0 µL Total volume 25 µL -
7. Perform FPCR using the following program.
Number of cycles Time Temperature 1 3 min 94°C 30 sec 55°C 1 min 72°C 25 30 sec 94°C 30 sec 55°C 1 min/kb 72°C 1 3 min 72°C Hold 4°C -
8. Run 5 µL of the FPCR product on a 1% agarose gel.
-
If the PCR was successful, a bright band of the size of the target synthetic DNA should be visible. Successful FPCR products are cloned into the pCR Blunt II-TOPO vector in the next section.
-
Cloning the Building Blocks
-
9. Assemble the following ligation reaction using the Zero Blunt cloning kit.
Reagent Volume FPCR product 0.5 µL Salt solution 0.5 µL pCR Blunt II-TOPO vector DNA 0.5 µL H2O (sterile) 1.5 µL Total volume 3 µL -
10. Mix the reaction well by pipetting up and down, and incubate for 5 min at room temperature.
-
11. Place the reaction on ice until ready to proceed with Step 13.
Transforming Bacteria
-
12. For each transformation, thaw one 25-µL aliquot of competent cells on ice.
-
13. Add 1 µL of the ligation reaction directly to each vial of cells and mix by stirring gently with a pipette tip.
-
14. Incubate the vials on ice for 5 min.
-
15. Heat shock the cells for 30 sec at 42°C. Immediately place the vials on ice for 2 min.
-
16. Add 125 µL of LB liquid medium to each vial.
-
17. Using glass beads, spread 125 µL from each vial on plates containing LB solid medium with kanamycin + X-gal.
-
18. Incubate the plates overnight at 30°C.
Performing Colony Screening PCR (csPCR)
-
19. Transfer individual white colonies from each transformation plate to sterile 96-well plates containing 100 µL of LB liquid medium with kanamycin per well.
-
We recommend picking six clones per transformation plate.
-
-
20. Incubate the liquid cultures overnight at 37°C.
-
21. For each csPCR, assemble the following reaction.
Reagent Volume 2× GoTaq Green 6.25 µL Forward primer (M13F) 0.25 µL Reverse primer (M13R) 0.25 µL Overnight bacterial culture 1 µL H2O (sterile) 4.75 µL Total volume 12.5 µL -
22. Perform csPCR using the following program.
Number of cycles Time Temperature 1 4 min 94°C 30 30 sec 94°C 30 sec 55°C 1 min/kb 72°C 1 3 min 72°C Hold 4°C -
23. Run 10 µL of csPCR product on a 1% agarose gel without loading dye.
-
If the PCR was successful, a bright band of roughly the size of the target sequence length should be visible.
-
-
24. Select csPCR-positive clones for sequencing with M13F and M13R.
-
Additional primers may be needed if the target sequence is longer than 1000 bp.
-
Assembling Building Blocks into Chunks
-
The above method is satisfactory for the synthesis of small building blocks (750 bp). However, to achieve high-efficiency integration and replacement of native chromosomes, much larger chunks of DNA are required. Similarly, many genes and pathways may contain longer DNA sequences. The following describes assembly of multiple building blocks into chunks using Gibson Assembly, the one-step isothermal DNA assembly method by Gibson et al. (2009).
-
25. Thaw 15 µL of isothermal reaction (Gibson Assembly) master mix on ice.
-
26. Combine all of the building blocks, vector backbone, and yeast selectable marker in equimolar amounts in a 5-µL volume.
-
27. Combine the building block mixture with 15 µL of isothermal reaction master mix in a PCR tube. Mix well and keep on ice.
-
28. Preheat the thermal cycler to 50°C.
-
29. Pause the thermal cycler at 50°C and transfer the PCR tube to the machine. Incubate the reaction for 30 min at 50°C.
-
30. Transform 2 µL of the assembly reaction into 50 µL of competent cells as described in Steps 14–18, plating the cells on the appropriate selective solid medium.
-
Successfully assembled chunks can be recovered from the bacteria and analyzed by restriction digestion or DNA sequencing.
-
DISCUSSION
Advances in high-throughput sequencing technologies (i.e., next-generation sequencing or NGS) have greatly enhanced our ability to read genetic information, and we are now able to read more than 15 petabases per year (Schatz and Phillippy 2012). However, our ability to write DNA is much more limited, especially at the genomic level. To date, only a few small genomes (from organisms such as poliovirus, bacteriophage, and mycoplasma) have been synthesized successfully (Cello et al. 2002; Smith et al. 2003; Gibson et al. 2008, 2010). Recently, we reported the synthesis of the first eukaryotic chromosome arms and a full yeast chromosome, beginning the journey toward a complete synthetic yeast genome (Dymond et al. 2011; Annaluru et al. 2014). In addition to complete genome synthesis, there is also a great need in the field of metabolic engineering for smaller synthetic genes and genetic pathways. Generally, synthesis of these DNA fragments can be outsourced to commercial vendors; however, despite continual drops in price, commercial gene synthesis remains too costly for many laboratories.
Several methods for gene synthesis have been developed in the past few decades. Currently, all gene synthesis technologies begin with chemical synthesis of short oligonucleotides (30–100 nt). The first-generation method relies on synthesis of oligonucleotides that completely cover both strands of DNA, which are phosphorylated and ligated in vitro. To save on the cost of oligonucleotide synthesis, researchers have also developed PCR-based methods, which require only 1.5× or lower coverage of a given DNA sequence, with no need for phosphorylated oligonucleotides. This is the method we adopted to synthesize the yeast genome, described in detail in this protocol. More recently, Gibson et al. (2009) developed an in vitro isothermal assembly method (now termed Gibson assembly), which allows small DNA fragments to be further assembled into 3 kb or larger fragments. Moreover, using the budding yeast as manufacturer, one can assemble DNA fragments from several kilobases to more than 1 million base pairs using in vivo recombination.
One difficulty of gene synthesis is the reduction of errors in the final products. These errors may come from the oligonucleotides or result from mistakes by DNA polymerase during PCR. To reduce error rates, oligonucleotide sizes must be limited for the current chemistry, or new chemistry must be developed. A few error correction techniques have been widely adopted in gene synthesis, such as the use of enzymes (Carr et al. 2004). To address this issue, we designed this protocol as a hierarchical procedure (Fig. 1). Building blocks (750 bp) are synthesized from overlapping oligonucleotides via high-fidelity PCR and then sequence verified to ensure 100% accuracy. These building blocks are designed to overlap for use with the Gibson Assembly method to form chunks (3–30 kb). Finally, each chunk is swapped into a yeast chromosome using in vivo homologous recombination. The two protocol sections presented here, beginning with overlapping oligonucleotides and covering building block synthesis through assembly of chunks ready for chromosome swap, are essentially independent. They can be used to synthesize DNA sequences of various sizes, and therefore they are not limited to the synthesis of DNA for the yeast genome.
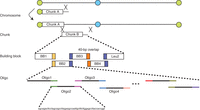
Hierarchy of DNA synthesis and assembly of a synthetic yeast chromosome.
ACKNOWLEDGMENTS
We thank Dr. Jef Boeke and Dr. Karen Zeller and the Build-a-Genome classes for their collective contribution to the protocol described here. Work in Y.C.’s laboratory is supported by a Chancellor’s Fellowship from the University of Edinburgh, a start-up fund from Scottish Universities Life Sciences Alliance, and Biotechnology and Biological Sciences Research Council (BBSRC) grant BB/M005690/1. Work in J.D.’s laboratory is supported by National Science Foundation of China 31471254 and 81171999, Chinese Minister of Science and Technology 2012CB725201, and Tsinghua University Initiative Scientific Research Program 20121087956.








