
Mapping of In Vivo RNA-Binding Sites by Ultraviolet (UV)-Cross-Linking Immunoprecipitation (CLIP)
Abstract
RNA “CLIP” (cross-linking immunoprecipitation), the method by which RNA–protein complexes are covalently cross-linked and purified and the RNA sequenced, has attracted attention as a powerful means of developing genome-wide maps of direct, functional RNA–protein interaction sites. These maps have been used to identify points of regulation, and they hold promise for understanding the dynamics of RNA regulation in normal cell function and its dysregulation in disease.
INTRODUCTION
Understanding how RNA is regulated opens the prospect of new insights into organismal complexity. Consider, for example, worms and humans—each very different, yet each harboring approximately the same number of classical protein-coding genes. The recognition of limits to DNA complexity, coupled with the excitement engendered by the concept of the RNA world and emerging evidence of great RNA complexity (see Cech 2009; Sharp 2009; Licatalosi and Darnell 2010), has suggested a solution to this conundrum. A modern view posits that the DNA in each organism is a “zip-file” of genetic information that is unfolded by a complex set of regulatory pathways into the RNA world, in which the diversity within each organism becomes manifest. In this view, understanding the regulation of RNA metabolism offers a new avenue toward understanding complex cellular-, tissue-, and organ-specific functions.
RNA CLIP (cross-linking immunoprecipitation) was originally developed in 2003 (Ule et al. 2003), and since then, its use has expanded greatly with the application of high-throughput sequencing methods (HITS-CLIP, also called CLIP-seq) to analyze the cross-linked RNA sequences (Licatalosi et al. 2008). By 2011, CLIP or HITS-CLIP had been used to analyze RNA–protein interactions in tissues and organisms as diverse as the eubacterium Deinococcus radiodurans, the filamentous fungus Ustilago maydis, Saccharomyces cerevisiae, Caenorhabditis elegans, HeLa and 293T cells, human embryonic stem cells, and mouse brain or seminiferous tubules (Table 1; Darnell 2010).
Representative RNA-binding proteins studied by CLIP
Cross-linking approaches to study RNA–protein interactions are analogous in many ways to chromatin immunoprecipitation (ChIP) techniques developed a decade or so earlier and used for analysis of DNA–protein interactions. Historically, the development of ChIP presaged CLIP, but the two techniques have now diverged in several significant ways. Both methods were developed to preserve native protein–nucleic acid interactions that might otherwise be lost or reassorted during purification, an issue recognized both for DNA–protein interactions (Kuo and Allis 1999) and RNA–protein interactions (Mili and Steitz 2004). Protein–DNA cross-linking methods were first developed in the 1980s in the analysis of transcription and chromatin regulatory factors (Ilyin and Georgiev 1969; Gilmour and Lis 1984; Solomon and Varshavsky 1985) and came into widespread use by the late 1990s with the advent of DNA ChIP, for example, in the study of histone–DNA interactions (Kuo and Allis 1999).
UV irradiation (Alexander and Moroson 1962; Smith 1962) and treatment with formaldehyde (Ilyin and Georgiev 1969) were both recognized early on to be able to cross-link DNA–protein complexes in vitro. In 1974, UV irradiation was also shown to be capable of cross-linking RNA to protein (Schoemaker and Schimmel 1974). By the mid-1980s, these techniques began to be applied in vivo (Gilmour and Lis 1984), for example, to analyze (Mayrand and Pederson 1981; Mayrand et al. 1981; Li et al. 2006), immunoprecipitate, and generate antibodies to ribonucleoprotein (RNP) complexes in irradiated cells (Dreyfuss et al. 1984). However, at that time, UV irradiation was not considered as a means to purify either protein-bound DNA or RNA. This possibility was overlooked for two reasons: a belief that UV-cross-linking efficiency would be too low to be useful (Fecko et al. 2007), and because UV cross-linking was already known to block reverse transcriptase (RT; used to map RNA–protein-binding sites) (Urlaub et al. 2002). Consequently, the reversibility of formaldehyde cross-linking (Kuo and Allis 1999) was an important factor in its incorporation into ChIP protocols.
Concerns about UV cross-linking were laid to rest in 2003 when the first CLIP experiments showed that the block of RT following UV cross-linking was itself inefficient, such that irradiated samples could, following proteinase K treatment, be reverse-transcribed and PCR-amplified, and products from the cross-linked RNA sequenced. The CLIP protocols established at that time form the basis, with some modifications, for the methods described in this introduction. However, the potential for a partial blockade of RT continues to generate some concern and has driven the development of alternative CLIP protocols (iCLIP) (Konig et al. 2010). Fortunately, this issue has not proven to be a barrier to identifying bound RNA fragments; the “standard” UV protocols described below are able to generate millions of unique RNA sequences (tags) cross-linked to many different RNA-binding proteins (RNABPs). Indeed, there are even advantages to the problems that RT encounters at sites of cross-linking: errors or partial RT arrest at cross-linking sites has been exploited to map the exact sites of RNA–protein interaction (see below) (Ule et al. 2005; Granneman et al. 2009; Hafner et al. 2010; Konig et al. 2010; Zhang et al. 2010; Zhang and Darnell 2011).
RNA CLIP using UV cross-linking offers distinct advantages over standard DNA ChIP using formaldehyde cross-linking. CLIP has the potential for higher temporal and spatial resolution because UV irradiation generates cross-linking only when the interaction between protein and nucleic acid is direct (within a bond length). This greater specificity of CLIP, discussed below, provides an important contrast to formaldehyde cross-linking, in which large protein–nucleic acid and protein–protein complexes become cross-linked.
Because UV cross-linking “freezes” RNA–protein interactions by generating a covalent bond, CLIP creates a snapshot of binding unaffected by later processing steps. This is a considerable advantage over methods that lack a cross-linking step, including the most common alternative, RNP immunoprecipitation (RIP). Critically, CLIP also identifies protein-binding sites within individual RNA molecules. When combined with high-throughput sequencing of cross-linked RNAs, CLIP generates genome-wide maps of protein–RNA-binding sites. Furthermore, HITS-CLIP has the capacity to generate predictive regulatory models for several RNA-binding proteins (Darnell 2010; Licatalosi and Darnell 2010; Darnell et al. 2011), including those involving multiple RNA–protein interactions, for example, those in Argonaute miRNA–mRNA complexes (Chi et al. 2009; Hafner et al. 2010; Zisoulis et al. 2010; Leung et al. 2011). HITS-CLIP offers the investigator an opportunity to integrate the activity of regulatory RNABPs with small RNA regulatory networks (see Box 1 and Box 2).
Mechanism and specificity of UV-protein cross-linking
The mechanism of UV-mediated cross-linking is not fully understood but is believed to be from absorption of UV light at 250–280 nm by nucleic acid bases (Brimacombe et al. 1988). This is thought to excite ground-state electrons to a singlet high-energy state, allowing them to form a new covalent bond with molecules that are in direct contact with the nucleotide (Fecko et al. 2007). Because cross-linking only occurs between molecules that are on the order of angstroms apart, in CLIP, only direct protein–RNA contacts are cross-linked and analyzed. UV irradiation does not induce protein–protein cross-links, although it generates DNA–DNA and RNA–RNA cross-links (Zwieb et al. 1978; Brimacombe et al. 1988), which have been used in a variety of biochemical contexts such as structural mapping of the ribosome.
Protein–RNA cross-linking reactions occur at only a minority of contact sites. (In an estimate done in our own studies using purified recombinant protein and a high-affinity RNA aptamer, maximal cross-linking efficiency plateaued between 1% and 5%, although this is likely to vary somewhat with different proteins [Fecko et al. 2007].) Notably, the specificity of UV cross-linking of proteins to RNA is different from that seen with formaldehyde cross-linking, which is used in DNA ChIP, and which has also been used to analyze protein–RNA interactions (Vasudevan and Steitz 2007; Yong et al. 2010). In general, formaldehyde cross-linking generates more extensive protein–nucleic acid and protein–protein complexes and entails temporal restrictions because the formaldehyde must penetrate tissues. These aspects of formaldehyde cross-linking may complicate genome-wide efforts to identify direct RNA–protein interaction sites.
The specificity of cross-linking between nucleic acid bases and amino acid side chains is incompletely understood at the biophysical level. There is a reported preference for UV to cross-link certain amino acids and nucleotides, particularly for thymidines in protein–DNA interactions studied with high-intensity lasers (a 8-nsec Nd:YAG laser at ∼100 MW cm2) (Hockensmith et al. 1986). However, this is not well established, in part because of the lack of consensus regarding either the means of assessing protein–nucleic acid cross-linking or conclusions regarding mechanism (Fecko et al. 2007). Studies of RNA–protein interaction have shown that UV cross-linking can induce covalent-bond formation between a large variety of amino acids and both purines and pyrimidines (Havron and Sperling 1977). For example, all 20 amino acids were found to cross-link to polyuridylic acid in vitro (Shetlar et al. 1984). On the basis of studies like these, it is generally believed that any amino acid is capable of being UV cross-linked to any nucleotide residue (Hockensmith et al. 1986). Consistent with these in vitro studies, CLIP has been used to identify specific binding sites for RNABPs with a wide variety of sequence preferences (YCAY, CU, U-rich, GA elements). Moreover, HITS-CLIP analysis of Argonaute proteins, which are targeted by miRNAs to specific sites but are able to cross-link surrounding mRNA sequences, do not show any nucleotide bias for the surrounding mRNA sequence (Chi et al. 2009; Hafner et al. 2010; Zisoulis et al. 2010).
In theory, given the advantages in precision of UV cross-linking relative to formaldehyde cross-linking, DNA CLIP (Law et al. 1998) may provide a higher-resolution means of assessing DNA–protein interactions than current ChIP-seq methods. However, DNA-CLIP may not be able to compete with the power and speed of current bioinformatic methods to resolve sites of DNA–protein interaction (Johnson et al. 2007).
HITS-CLIP data analysis
Combining HITS-CLIP with complementary approaches
HITS-CLIP allows a genome-wide assessment of interaction sites. One principle that has emerged from these studies is that the analysis of such binding sites is in many cases most productive when combined with genome-wide functional analyses of RNA variants. Examples include microarray or RNA-seq studies of RNA variations detectable as a function of some perturbation, often a genetic perturbation (knockdown or genetic null). Not surprisingly, given the large amounts of data generated in such experiments, an important third leg of such analyses is a bioinformatic/computational approach. A general discussion of the combination of these points in the analysis of HITS-CLIP data has been recently reviewed (Licatalosi and Darnell 2010).
One informative example of the combined use of functional analyses came from a reexamination of HITS-CLIP data (Xue et al. 2009) in light of a genome-wide analysis of polypyrimidine tract-binding protein (PTB)-dependent splice variants (Llorian et al. 2010). This combination of approaches, combined with bioinformatic analysis, allowed reinterpretation of the CLIP data to unmask an RNA regulatory map in which the position of PTB binding determined the outcome of splicing inhibition or enhancement (Llorian et al. 2010) in a manner analogous to that seen for several other RNABPs, including Nova (Ule et al. 2006), Fox2 (Zhang et al. 2008; Yeo et al. 2009), heterogeneous nuclear ribonucleoprotein (hnRNP) L (Hung et al. 2008), hnRNP C (Konig et al. 2010), Muscleblind-like protein (MBNL) (Du et al. 2010), and TIA1/L (Wang et al. 2010).
Bioinformatic and Computational Analysis of HITS-CLIP Data
Examples have been published on the use of bioinformatics to delineate binding sites and clusters of CLIP tags to define binding footprints (Licatalosi et al. 2008; Granneman et al. 2009; Sanford et al. 2009; Wang et al. 2010; Khorshid et al. 2011; Kishore et al. 2011), to use Argonaute mRNA footprints to predict miRNA-binding sites (Licatalosi et al. 2008; Granneman et al. 2009; Sanford et al. 2009; Wang et al. 2010), or to develop predictive computational tools (Zhang et al. 2010).
Some general points of strategy are worth delineating. The basic bioinformatic pipeline for dealing with large amounts of next-generation sequence data consists of several sections. First, raw tags, typically ∼20–50 nt or longer, are bioinformatically trimmed of any linker/adaptor sequences and then mapped to the genome. Although saving only tags that map uniquely to the genome is a useful general strategy, one must be aware that pseudogenes or repetitive genes (such as ribosomal RNAs) may eliminate bona fide tags that appear to map to more than one genomic locus. Most commonly, multiple representations of a single RNA tag are collapsed to a more refined set of unique sequences. This is done to eliminate overamplification bias from PCR amplification steps. In the future, direct RNA sequencing applied to the analysis of HITS-CLIP tags may eliminate such concerns. In the meantime, in protocols that use RT-PCR, bar-coded linkers can help to discriminate PCR duplicates from true unique RNA tags (Konig et al. 2010). Similarly, if contamination may be an issue (especially when multiple CLIP experiments are planned), in addition to standard PCR precautions, the use of indexed linkers (Cronn et al. 2008) may be considered, enabling each experiment to have a unique nucleotide code.
With a set of winnowed RNA tags, unique in sequence and mapped to the genome, higher-order analysis of data is possible. RNA-binding proteins are likely to have legitimate transit times on illegitimate (nonfunctional) RNA sequences, and such physiological background, in addition to biochemical background tags, often needs to be eliminated. One way that this may be done is by normalizing data to random expected tags per transcript, as was done in mapping Argonaute mRNA footprints (Chi et al. 2009). Another general approach is to focus on overlapping RNA tags because these define reproducible sites of RNA–protein interaction; in complex biological experiments, we have found it useful to focus on tags that are present in more than one biologic replicate experiment (defined as loci with a “biologic complexity” of >n, where n is the number of replicate experiments) (Licatalosi et al. 2008). Such loci may be further refined by making a minimal threshold for the number of tags per cluster (peak height). Typically, such analysis of HITS-CLIP data is not difficult and can be readily accomplished even with large data sets using a program like Excel.
Recent bioinformatic studies have refined RNA–protein mapping obtained from the standard HITS-CLIP protocol to single-nucleotide resolution. This was initially thought to be an advantage specific to PAR-CLIP, which analyzes mutations induced by nucleotide analogs and UV irradiation to map cross-linking sites. However, it was noticed early on in standard CLIP (Ule et al. 2005; Granneman et al. 2009) that there were an increased number of mutations in specific locations within cross-link clusters. This was formalized computationally and established bioinformatically for Nova and Argonaut HITS-CLIP experiments with cross-link-induced mutation (CIMS) analysis (Zhang and Darnell 2011), which uses these mutations to map cross-linked nucleotides. This approach does not require the use of nucleotide analogs (i.e., can be performed with native cells or tissues) and thus offers a significant advantage over PAR-CLIP (Kishore et al. 2011).
It is now standard practice to deposit all of the CLIP tags obtained from a HITS-CLIP experiment, at the time of publication, into a publically accessible site such as that maintained by the National Institutes of Health (the Gene Expression Omnibus [GEO]; http://www.ncbi.nlm.nih.gov/geo/).
THE CROSS-LINKING IMMUNOPRECIPITATION METHOD
The cross-linking immunoprecipitation (CLIP) method (Fig. 1) provides a general approach to mapping RNA–protein interactions in vivo, whether in whole tissues, organisms, or individual cell types treated with UV irradiation at ∼254 nm (Ule et al. 2003, 2005; Jensen and Darnell 2008). This treatment induces a transient excited electron state that generates a covalent bond between RNA and protein molecules that are in very close contact (at most several angstroms apart). Although the exact mechanism is not fully understood, it is believed that the absorption of UV light by nucleic acid bases (Brimacombe et al. 1988) induces ground-state electrons to a singlet higher-energy state, enabling the electron to form a new covalent bond (Fecko et al. 2007). The protein–RNA cross-linking reaction occurs at only a minority of contact sites (∼1%–5% in standard CLIP experiments, although this may vary with different proteins) (see also Fecko et al. 2007). (See also Box 1.)
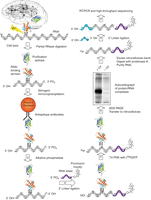
Cross-linking immunoprecipitation (CLIP). Tissue (e.g., brain) or cells are UV-irradiated (see Protocol: Ultraviolet (UV) Cross-Linking of Live Cells, Lysate Preparation, and RNase Titration for Cross-Linking Immunoprecipitation (CLIP) [Darnell et al. 2018a]), inducing covalent cross-links between RNA–protein complexes in vivo. (Arrow 1, see Protocol: Ultraviolet (UV) Cross-Linking of Live Cells, Lysate Preparation, and RNase Titration for Cross-Linking Immunoprecipitation (CLIP) [Darnell et al. 2018a] and Protocol: Immunoprecipitation and SDS-PAGE for Cross-Linking Immunoprecipitation (CLIP) [Darnell et al. 2018b]) Cell lysis and partial RNase digestion allow partial clarification of RNA–protein complexes before immunoprecipitation and reduce the modal size of cross-linked RNA to a size determined by the experimenter (in typical experiments, this would be ∼50 nt or smaller). RNase A and T1 leave a 5′-OH and a 3′-phosphate group on the digested RNA. (Arrow 2, see Protocol: Immunoprecipitation and SDS-PAGE for CLIP [Darnell et al. 2018b]) Cross-linking allows stringent conditions to be used for protein purification. Immunopurification using antibodies against native epitopes or protein tags can be used. (Arrows 3, 4, see Protocol: 3′-Linker Ligation and Size Selection by SDS-PAGE for Cross-Linking Immunoprecipitation (CLIP) [Darnell et al. 2018c]) The 3′ phosphate is removed by alkaline phosphatase, to prevent intramolecular RNA circularization during ligation of a linker to the 3′ end of the RNA. This linker is itself blocked at the 3′ end with a puromycin moiety to prevent competing linker–linker ligation reactions. (Arrow 5, see Protocol: 3′-Linker Ligation and Size Selection by SDS-PAGE for Cross-Linking Immunoprecipitation (CLIP) [Darnell et al. 2018c]) RNA is labeled at the 5′ end with T4 polynucleotide kinase and [γ-32P]ATP. (Arrow 6, see Protocol: 3′-Linker Ligation and Size Selection by SDS-PAGE for (CLIP) [Darnell et al. 2018c]) RNABP:RNA complexes are released from beads, run on denaturing SDS-PAGE and transferred to nitrocellulose (two important purification steps), and imaged by autoradiography. (Arrow 7, see Protocol: Isolation of the RNA Cross-Linking Immunoprecipitation (CLIP) Tags, 5′-Linker Ligation, Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Amplification, and Sequencing [Darnell et al. 2018d]) The radioactive RNABP–protein complex is excised from the nitrocellulose filter and digested with proteinase K to remove the RNABP and elute the RNA, which is then isolated by phenol:chloroform extraction and ethanol precipitation. (Arrow 8, see Protocol: Isolation of the RNA Cross-Linking Immunoprecipitation (CLIP) Tags, 5′-Linker Ligation, Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Amplification, and Sequencing [Darnell et al. 2018d]) A second linker is added to the 5′ end of the RNA. (Arrow 9, see Protocol: Isolation of the RNA Cross-Linking Immunoprecipitation (CLIP) Tags, 5′-Linker Ligation, Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Amplification, and Sequencing [Darnell et al. 2018d]) The RNA is amplified by RT-PCR and sequenced.
Following formation of this new bond, the RNA–protein complex can be purified under very stringent conditions. In principle, any purification method can be used. But in practice, the foundations of a robust protocol are built from three steps: (1) immunoprecipitation with antibodies to the RNA-binding proteins (RNABPs) themselves or to transgenic epitope tags, (2) size separation by SDS–PAGE, and (3) transfer to nitrocellulose to remove contaminating free (non-cross-linked) RNA. In the course of this purification, RNA is intentionally reduced in size, typically to a modal size of ∼50 nt, to facilitate identification of binding sites (e.g., cross-linked RNAs from ∼20 to 100 nt). Once sufficient purity has been obtained, the protein component of the cross-linked complex is removed with proteinase K treatment, and the released RNA is purified. Current protocols, which are still evolving, use RNA ligase to attach RNA linkers to the released RNA pool; templates for sequencing are generated by cDNA synthesis using an antisense primer and reverse transcriptase.
One of the main advantages of CLIP is that it can be performed in vivo in many different systems. Although CLIP was first undertaken in mouse brain, it has since been applied to a range of whole organisms, including bacteria, fungi, yeast, C. elegans, and several mammalian tissue culture cells including human embryonic stem cells (Table 1). CLIP has also been applied to a number of different RNA-binding proteins involved in an array of different biological processes (Table 1). For example, even the small number of CLIP sequence tags derived for heterogeneous nuclear ribonucleoprotein (hnRNP) A1 from tissue culture cells was sufficient to suggest that this complex might regulate pre-miRNA processing (Guil and Caceres 2007), an observation subsequently pursued mechanistically (Michlewski et al. 2008) and one that may have more general implications for microRNA (miRNA) regulation (Newman et al. 2008). Analysis of a small number of Nova (a 55-kDa RNABP found in the nucleus and cytoplasm of mouse brain neurons) CLIP tags revealed sites of functionally relevant RNA–protein interactions (Ule et al. 2003) whose significance was subsequently confirmed in higher-throughput studies (Licatalosi et al. 2008).
HIGH-THROUGHPUT SEQUENCING (HITS) CLIP
When CLIP was first developed, 340 unique Nova-bound RNAs were sequenced at a cost of ∼$4000 (Ule et al. 2003). The small number of tags precluded robust generalizations about the nature of Nova’s interaction with RNA. These studies were extended by applying high-throughput sequencing methods to CLIP, termed HITS-CLIP (or CLIP-seq). Reanalysis of the same RNA–protein interactions with HITS-CLIP in 2008 identified 1000-fold more unique tags for the same cost of ∼$4000. The price per RNA tag continues to drop dramatically. Given such large data sets, analyzing RNA CLIP tags using bioinformatic methods becomes an important component of any HITS-CLIP study.
RNA–protein maps obtained with HITS-CLIP can be combined with the results of other studies to reveal correlations between binding position and function of proteins on RNA. For example, together with bioinformatic studies (Ule et al. 2006), RNA–protein maps reveal that the position of binding determines the outcome of Nova-mediated splicing regulation (exon inclusion/exclusion) (Licatalosi et al. 2008). Subsequent studies, including HITS-CLIP with PTB (Xue et al. 2009) combined with the functional analysis of PTB-dependent RNA splicing variants (Llorian et al. 2010), and additional studies with other RNABPs—Fox2, hnRNP C, hnRNPL, TIA1/2, TDP-43, MBNL, and CELF proteins (Yuan et al. 2007; Kalsotra et al. 2008; Zhang et al. 2008; Yeo et al. 2009; Du et al. 2010) and other RNABPs (Chen and Manley 2009; Tollervey et al. 2011; Witten and Ule 2011)—have led to the recognition that such position-dependent splicing maps are a general phenomenon. They reveal rules of splicing regulation that are common to many RNA-binding proteins (Chen and Manley 2009; Corrionero and Valcarcel 2009; Licatalosi and Darnell 2010; Witten and Ule 2011). HITS-CLIP also uncovered a role for Nova in the regulation of alternative polyadenylation (Licatalosi et al. 2008). HITS-CLIP studies with hnRNP C suggest that the protein may form higher-order RNA–protein complexes, perhaps analogous to DNA nucleosomes, and thereby play an important role in splicing inhibition (Konig et al. 2010). Finally, genome-wide mapping of Argonaute (Ago) footprints with HITS-CLIP identified many binding sites outside of the 3′ untranslated region (3′ UTR), suggesting new points of Ago-miRNA regulation of RNA transcripts (Chi et al. 2009; Hafner et al. 2010; Zisoulis et al. 2010). Such uses of HITS-CLIP provide a new adjunct to traditional approaches to studying regulatory mechanisms (Sharp 2009; Xue et al. 2009; Nilsen and Graveley 2010).
VALIDATION OF CLIP RESULTS
To generate functional RNA–protein maps, it is desirable to correlate the species of physical RNA–protein interactions generated by CLIP with the identification of RNA variants, detected, for example, by RNA-seq analysis in different tissues or genetic backgrounds, and with bioinformatic analyses, which together can provide a general approach to developing functional RNA–protein maps (Licatalosi and Darnell 2010). Often these parallel experiments synergize with CLIP data to produce new biological insights. Examples include combining CLIP data with independent RNA microarray analyses (Ule et al. 2005; Licatalosi et al. 2008; Daughters et al. 2009; Yeo et al. 2009), RNA sequence analysis (Bohnsack et al. 2009; Chi et al. 2009; Hafner et al. 2010; Llorian et al. 2010; Zisoulis et al. 2010), functional assays of RNA–protein interactions (Chi et al. 2009; Darnell et al. 2011), physiology (Huang et al. 2005; Ruggiu et al. 2009), or even cellular studies of RNA localization (Racca et al. 2010) or cell migration (Yano et al. 2010).
Cross-validation comparing CLIP data with other types of data includes studies of Fox2 (Yeo et al. 2009) with data independently confirmed bioinformatically by Zhang et al. (2008) and of PTB (Xue et al. 2009) with data independently assessed biochemically and bioinformatically (Gama-Carvalho et al. 2006; Boutz et al. 2007; Xing et al. 2008; Llorian et al. 2010). Examples of independent confirmation of CLIP studies also include those independently performed by different laboratories, as in HITS-CLIP studies of Ago (Chi et al. 2009; Hafner et al. 2010; Zisoulis et al. 2010).
CLIP METHOD VARIATIONS
Variations of the basic CLIP methods have been and are likely to continue to be developed (Table 2). These methods share common features: UV irradiation to cross-link RNA–protein complexes within living cells, followed by stringent conditions to purify RNA–protein complexes, partial digestion of RNA to produce clonable fragments, and linker or adaptor ligation to allow RT-PCR amplification and sequencing of cDNA (or, in emerging methodology, allowing direct sequencing of RNA) (Ozsolak et al. 2009). One variation describes methods to tag proteins before CLIP to allow protein purification (“cross-linking reactions and purification, or cross-linking and analyses of cDNA [CRAC]”) (Granneman et al. 2009). The introduction of tagged proteins as in CRAC may prove useful where other means of purifying RNA-binding proteins are not feasible, although attention to stoichiometry is critical if the goal is the identification of biologically relevant sites. Methods to identify sites of cross-linking have been described, including monitoring reverse transcriptase pausing (individual-nucleotide resolution UV cross-linking and immunoprecipitation [iCLIP]) (Konig et al. 2010) and identifying nucleotide changes following incorporation of 4-thiouridine into RNA in tissue culture cells (photoactivatable-ribonucleoside-enhanced cross-linking and immunoprecipitation [PAR-CLIP]) (Hafner et al. 2010). Such information can also be obtained by bioinformatic analysis of sequence errors or pause sites introduced by RT in the basic CLIP protocol (Ule et al. 2005; Granneman et al. 2009; Konig et al. 2010; Zhang and Darnell 2011).
Variant CLIP protocols
CLIP technology is relatively new and is evolving through the work of many laboratories (Darnell 2010). For many applications, the generation of millions of CLIP tags with the basic CLIP protocol outlined here is sufficient to generate a bioinformatically winnowed set of hundreds of thousands of unique CLIP tags, which can then be used to generate informative genome-wide RNA maps.
GENERAL CONSIDERATIONS IN PLANNING CLIP EXPERIMENTS
There are several issues to consider before beginning a CLIP experiment on an RNA-binding protein of interest. A key decision is the choice of starting material. One of the main advantages of CLIP is that it allows the detection of physiologically relevant interactions by using UV on intact cells or living tissues to take a snapshot in time of RNABP:RNA binding. In consequence, there is great choice of organism, tissue, developmental stage, and control over perturbations such as cellular or neuronal activity, and biologically relevant questions can and should be chosen with care. Another critical factor is the successful purification of the RNABP:RNA complex after cross-linking—traditionally one step in the process is an immunoprecipitation. Because RNA–protein interactions are sensitive to stoichiometry (i.e., excess protein may lead to nonspecific RNA interaction), we advocate the use of antibodies to endogenous proteins wherever possible, to favor identification of physiologically relevant RNA–protein interactions. In cases in which this is not practical, transgenic expression of tagged versions of RNA-binding proteins may be considered. In these instances, too, it is important to use expression levels that are as close to physiological as possible, and thus knockin of a tag into an endogenous locus would be preferable to expression of a cDNA transgene.
Because the basic CLIP technique relies on immunoprecipitation as a major purification step (although, in theory, any means of protein purification could be used), the quality of the immunoprecipitation is frequently the rate-limiting factor in any CLIP experiment. An associated protocol (see Protocol: Immunoprecipitation and SDS-PAGE for Cross-Linking Immunoprecipitation (CLIP) [Darnell et al. 2018b]) provides a pilot experiment needed to evaluate the sensitivity and specificity of the planned CLIP experiment. As a rule, CLIP succeeds by taking advantage of the covalent bond to permit stringent immunoprecipitation and wash conditions to be used because there is no longer an issue of loss of the RNABP:RNA interaction, although the strength of the antibody:RNABP interaction must always be considered. As described in the associated protocols listed in the following paragraph, using increasingly stringent wash conditions is well worth the investigator’s time for evaluating the maximum stringency that can be used to detect the desired RNA–protein complexes.
Protocol: Ultraviolet (UV) Cross-Linking of Live Cells, Lysate Preparation, and RNase Titration for Cross-Linking Immunoprecipitation (CLIP) [Darnell et al. 2018a], Protocol: Immunoprecipitation and SDS-PAGE for Cross-Linking Immunoprecipitation (CLIP) [Darnell et al. 2018b], Protocol: 3′-Linker Ligation and Size Selection by SDS-PAGE for Cross-Linking Immunoprecipitation (CLIP) [Darnell et al. 2018c], and Protocol: Isolation of the RNA Cross-Linking Immunoprecipitation (CLIP) Tags, 5′-Linker Ligation, Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Amplification, and Sequencing [Darnell et al. 2018d] comprise a typical CLIP experiment. There is a natural stopping point between Protocol: 3′-Linker Ligation and Size Selection by SDS-PAGE for Cross-Linking Immunoprecipitation (CLIP) [Darnell et al. 2018c] and Protocol: Isolation of the RNA Cross-Linking Immunoprecipitation (CLIP) Tags, 5′-Linker Ligation, Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Amplification, and Sequencing [Darnell et al. 2018d] because of the time required to expose and analyze the RNABP:RNA complexes on the nitrocellulose filter. Conveniently, the filter can be frozen at this point at −80°C indefinitely and bands excised in the future. Following exposure and analysis of the results from Protocol: 3′-Linker Ligation and Size Selection by SDS-PAGE for Cross-Linking Immunoprecipitation (CLIP) [Darnell et al. 2018c], it may be evident that the experiment must be repeated from the beginning with increased starting material or with alterations to the purification steps in some manner. A full CLIP experiment can be performed within ∼1 wk once optimization of parameters (see Protocol: Immunoprecipitation and SDS-PAGE for Cross-Linking Immunoprecipitation (CLIP) [Darnell et al. 2018b]) has been completed. Subsequent RNA sequencing (see Protocol: Isolation of the RNA Cross-Linking Immunoprecipitation (CLIP) Tags, 5′-Linker Ligation, Reverse Transcription-Polymerase Chain Reaction (RT-PCR) Amplification, and Sequencing [Darnell et al. 2018d]) and bioinformatic analysis complete the HITS-CLIP experiment.
ACKNOWLEDGMENTS
We thank past and present members of the Robert Darnell laboratory for initial development and application of the CLIP method (especially Kirk Jensen and Jernej Ule), and application of high-throughput sequencing technologies (especially Donny Licatalosi, Sung Wook Chi, and Chaolin Zhang). We thank laboratory members for their continual contributions to protocol development and its applications to multiple RNA-binding proteins, as well as critical reading of the protocols presented. In addition, we thank all the investigators who have shared their own experiences using CLIP; protocols are maintained and questions and comments are welcome on our CLIP forum (http://lab.rockefeller.edu/darnell/).
Footnotes
-
From the Molecular Cloning collection, edited by Michael R. Green and Joseph Sambrook.








