
A Guide to Cloning the Products of Polymerase Chain Reactions
Abstract
This introduction outlines various methods to clone amplified DNAs and to facilitate the construction of complex multicomponent genetic units. Because of the ease with which the termini of amplified DNAs can be tailored by polymerase chain reaction (PCR), many of the methods outlined here use PCR not only to synthesize DNAs but also to link them together into purpose-designed constructs. The most recent refinements however have been the development of modular genetic units that can be harnessed to target DNAs not by PCR but by site-specific recombination enzymes.
BACKGROUND
Since the first cloning papers were published in the early 1970s, hundreds of different methods have been published describing how best to generate and assemble DNA constructs of increasing complexity and sophistication. In the days before PCR, these methods—of necessity—were permutations and modifications of just two enzymatic reactions: restriction and ligation. Despite the inventiveness of the early researchers, precise assembly of complex DNA structures remained a significant challenge. Hardly surprising then, that only one or two of the cloning methods that were so carefully developed in those early days remain in common use today. Most were simply swept aside in the late 1980s by the liberating tide of PCR. During the course of 25 years, an expanding repertoire of PCR techniques has come to dominate molecular cloning.
The following guide outlines various methods used to clone amplified DNAs and to facilitate the construction of complex multicomponent genetic units (for an overview of standard methods, see Table 1). [Note that the names and products of the manufacturers mentioned below were current at the time of this initial writing.]
Standard methods for cloning amplified DNA
SITE-SPECIFIC RECOMBINATION METHODS
In the last few years, cloning and subcloning of PCR products and other DNAs into prokaryotic vectors has undergone a revolution. No longer is it necessary to depend on ligation of compatible termini or on complex PCR protocols. Instead, DNA segments flanked by the appropriate recombination sites can be joined in vitro to vectors carrying compatible recombination sites through the action of sequence-specific recombinases such as bacteriophage λ integrase.
Recombinational cloning has several advantages over more traditional methods of cloning that rely on the restriction enzymes and ligases. Recombinational cloning:
-
is flexible and highly efficient
-
enables DNA fragments to be moved easily and rapidly from one expression vector to another in a single-step procedure that maintains orientation and reading frame
-
facilitates protein expression and functional analysis
-
can be adapted to high-throughput, automated formats
-
has produced large archives of clones, carrying mammalian open reading frames generated by the Gateway cloning system, that are publicly available from the Mammalian Gene Collection at NIH (http://mgc.nci.nih.gov/)
Originally developed for large-scale proteomic analyses, recombinational cloning is increasingly used for small-scale research projects and for most large-scale proteomic analyses. The two most popular versions of recombinational cloning—Gateway cloning (Life Technologies) and In-Fusion (Clontech)—use different sequence-specific recombinases and have different strengths and weaknesses. Both systems are patent protected.
-
Gateway cloning (Life Technologies) uses an in vitro version of the site-specific recombinational system used by bacteriophage λ DNA during integration into and excision from the E. coli genome (Hartley et al. 2000; Walhout et al. 2000; see also Introduction: Gateway Recombinational Cloning [Reece-Hoyes and Walhout 2018]). Three enzymes are involved: integrase (Int), excisionase (Xis), and integration host factor (IHF). These enzymes catalyze recombination at specific DNA sequences known genetically as attB, attP, attL, and attR. The Gateway system uses modified versions of these sequences and their cognate recombinases that increase the specificity of the recombination reaction without reducing its efficiency (see Table 2).
In Gateway cloning, a DNA fragment carrying attB1 at one end and attB2 at the other is prepared by PCR and cloned into a vector (the donor vector) carrying attP1 and attP2 recombination sites flanking the ccdB gene, a death gene in E. coli (for information about the ccdB gene and its protein, see Box 1). Cloning is performed in an in vitro recombination reaction, driven by a proprietary enzyme (BP clonase), during which the incoming DNA fragment replaces the ccdB sequences. The resulting recombinant plasmid is called an Entry clone and its flanking recombination sequences are called Gateway attL-type.
The DNA sequence carried by the Entry clone can then be transferred into any Destination vector that carries attR recombination sequences—a reaction that is catalyzed by LR Clonase—a proprietary mixture of enzymes. The power of the Gateway system is that it allows DNA fragments flanked by appropriate sequence-specific recombination sites to be transferred into a Destination vector in a highly efficient single-step procedure. Thousands of Destination vectors are available, equipped with prokaryotic, mammalian and viral promoters, signal sequences, splicing signals, Shine–Dalgarno sequences, cleavage sequences, poly(A) addition sequences, and many other elements required for (i) the expression in various types of cells and (ii) purification of the target protein. For further details on Gateway cloning, see Introduction: Gateway Recombinational Cloning (Reece-Hoyes and Walhout 2018) and Figures 1, 2, and 3 therein.
-
In-Fusion cloning uses a proprietary version of vaccinia virus-encoded DNA polymerase called the In-Fusion enzyme. This enzyme has three catalytic functions—strand displacement, exonucleolytic removal of nucleotides, and homologous recombination (Marsischky and LaBaer 2004; Hamilton et al. 2007). In reactions containing two double-stranded linear DNA substrates the 3′–5′ proofreading activity of the poxvirus DNA polymerase removes nucleotides from the 3′ termini of the DNAs. If the exposed single-stranded regions of the two molecules are homologous in sequence, they anneal to form a best-fit hybrid that is relatively resistant to further exonucleolytic attack by the polymerase. Any nicks, short overhangs, and gaps are repaired after the annealed DNAs are introduced into E. coli, using a standard transformation protocol.
In the In-Fusion system, the regions of homology are generated by adding to the forward and reverse PCR primers 15-base extensions that exactly match the known sequences at the ends of a linearized plasmid vector. The In-Fusion system can therefore be used to create circular recombinant plasmids from any DNA fragment whose terminal sequences have been appropriately tailored by PCR (Benoit et al. 2006; Zhu et al. 2007).
Clontech markets plasmid vectors that can be used to create Master clones in which the segment of cloned DNA is flanked by loxP sites (see Fig. 1). The sequences inserted into a Master clone can then be to any Acceptor vector that also carries a loxP site in an in vitro reaction catalyzed by Cre recombinase. A collection of Acceptor vectors engineered to express proteins in bacteria, yeast, and mammalian cells is available from Clontech. Like Gateway's Destination vectors, In-Fusion Expression vectors are equipped with a variety of signals, tags, and selective markers.
In summary, the In-Fusion system is simple, efficient, and rapid and can be adapted for high-throughput projects (e.g., the BioBricks project).
THE ccdB GENE
The ccdB gene, carried by the F plasmid of E. coli, encodes a protein of 101 amino acids residues that is lethal to bacteria in the absence of the antidote ccdA protein. Although both proteins are required for stable maintenance of the F plasmid, the half-life of the ccdA protein is shorter than that of ccdB. Because the copy number of the plasmid is low (one to two copies per cell), one or other of the two daughter cells may fail to inherit the F plasmid at division. As the level of ccdA protein drops, the F segregants become vulnerable to killing by the ccdB protein—a process known as postsegregational killing. Under natural conditions, the combined action of the two proteins encoded by the ccd locus ensures stable maintenance of the F plasmid in the bacterial population.
The ccdB protein kills cells by inducing ATP-dependent cleavage of DNA by gyrase, with resulting activation of the SOS system (Bernard and Couturier 1992; Miki et al. 1992; Bernard et al. 1993). However, the lethal effects of the protein are suppressed in strains of E. coli carrying the gyrA462 mutation, which enables high-copy-number plasmid vectors (e.g., pKIL; Bernard et al. 1994) carrying the ccdB gene to be maintained and grown to high copy number. If the ccdB gene in a pKIL vector is inactivated by insertion of a segment of foreign DNA into a multiple cloning site within the ccdB gene, the resulting recombinants become able to transform gyrA+ strains of E. coli. In the Gateway cloning system, the ccdB gene in the Destination vector is flanked by attR recombination sites. Cloning is performed by an in vitro recombination reaction during which an incoming DNA fragment replaces the toxic ccdB gene, allowing selective propagation of recombinants in gyrA+ cells (Hartley et al. 2000).
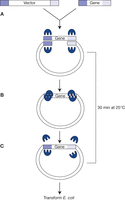
Ligation-independent cloning using In-Fusion (Clontech). (A) The reaction mixture contains a vector of choice linearized by a restriction cut, a PCR product generated with primers containing 15-bp 5′ ends homologous to the ends of the linear vector (gene), and the proprietary In-Fusion enzyme. (B) The enzyme catalyzes the alignment and strand displacement of the homologous ends of the PCR product with the vector, whereas 3′-exonuclease activity removes single-stranded regions. (C) The nicks are sealed after transformation of E. coli. (Reproduced from D'Arpa 2009.)
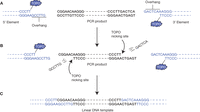
TOPO tools (PCMV/TetO 5′ element) for producing linear DNA constructs with defined 5′ and 3′ functional elements. (A) The 5′ and 3′ functional elements and the PCR product to which they are to be added are shown. Different 11-bp sequences (bold) are added to the ends of the PCR product via forward and reverse primers. (B) TOPO cleaves the PCR product, creating TOPO-activated overhangs (and the release of single-stranded 6-mers) complementary to the overhangs of the TOPO-activated functional elements. (C) Annealing the overhangs juxtaposes the 5′-OH of each to attack the phosphotyrosyl bond of the other, ligating both strands of each functional element to both strands of the PCR product, releasing two TOPO molecules, and creating a linear recombinant template for direct use in vitro and in vivo. (Reproduced from Invitrogen 2002, part of Life Technologies, 2002.)
Properties of the Gateway-modified att sites
Choosing between the Gateway and In-Fusion Systems
If the goal is to create individual clones or plasmid libraries, In-Fusion is the better choice, simply because it can be used with any plasmid vector whose DNA sequence is known. However, if the goal is to create libraries of Master clones (In-Fusion) or Entry clones (Gateway), the choice between the two systems is more difficult. Both are fully optimized, off-the-shelf, amenable to automation and easily adapted to high-throughput sequencing. In the only head-to-head comparison so far published (Marsischky and LaBaer 2004), the efficiencies of the two systems were similar, although In-Fusion was better able to handle cloned open reading frames larger than 2–3 kb. However, Gateway currently has the more comprehensive set of Destination vectors and these are freely shared among researchers. In addition, the Gateway system in combination with other recombineering techniques can be used to analyze and modify patterns of gene expression (Rozwadowski et al. 2008). In-Fusion, on the other hand, can be used for simultaneous and ordered assembly of several DNA fragments into a single construct in a technique called gene assembly (Sleight et al. 2010). Unfortunately, neither system is cheap. So before beginning any large-scale project it is essential to find out what clones/libraries are already available and to make a head-to-head comparison of the costs of the two systems and then develop a convincing business case.
OTHER CLONING METHODS
Ligation-Independent Cloning
Ligation-independent cloning (LIC-PCR) (also known as enzyme-free cloning) increases both the efficiency and speed of cloning of PCR products. LIC-PCR eliminates the need to ligate PCR products to a vector, does not rely on restriction sites, and avoids problems caused by extra bases that are added by the extendase activity of thermostable DNA polymerases at the 3′ termini of PCR products (Clark 1988). By allowing direct cloning of a PCR product into a particular site of a plasmid vector, LIC-PCR eliminates complex constructions that arise when a vector lacks suitable cloning sites, or a genetic basis for screening for recombinants (Aslanidis and de Jong 1990; Hsiao 1993; Yang et al. 1993; Kaluz and Flint 1994). Several variants of LIC-PCR have been described.
-
Amplification of the target with primers that carry at least 24 extra bases at their 5′ end. These bases correspond to sequences at the end of a linearized plasmid vector. Amplification with these primers therefore generates PCR products whose 5′ ends are complementary to the 3′ ends of the recipient linearized plasmid. The PCR product and the linearized plasmid are then spliced together in a second PCR, which extends the overlapping complementary 3′ ends (Shuldiner et al. 1990).
-
Generation of PCR products that carry 3′-protruding termini, 12 or more bases in length, that are annealed to complementary single-stranded sequences at the 3′ termini of a linearized vector. Base pairing between the two sets of protruding tails creates a chimeric molecule that can be introduced into E. coli by transformation (Aslanidis and de Jong 1990).
Methods to generate complementary single-stranded tails at the 3′ termini of the vector and the PCR product include exonuclease resection of 3′ termini. The sequences of the vector and the PCR product are designed so that one of the four bases does not occur within the first 12 nucleotides of the end of the 3′ strand. Complementary tails of defined length can therefore be produced by the 3′–5′ exonuclease activity of T4 DNA polymerase in the presence of a single dNTP. The enzyme removes nucleotides in the 3′–5′ direction until it reaches the first nucleotide that corresponds to the dNTP included in the reaction mix. Further exonucleolytic digestion by T4 DNA polymerase is neutralized by incorporation of the dNTP at the 3′-hydroxyl terminus of the recessed strand. After the enzyme has been inactivated by heat, the PCR product is annealed to a vector with a single-stranded tail prepared in a similar way (Aslanidis and de Jong 1990; Haun et al. 1992; Tachibana et al. 2009). When the chimeric molecule is used to transform E. coli, the ligase of the bacterial cells seals the single-stranded nicks and generates a covalently closed circular molecule.
In a variation of this method, overlapping sequences are designed into the PCR primers used to amplify the target DNA and the vector. Controlled digestion of the PCR product and the vector with either exonuclease III (Hsiao 1993; Li and Evans 1997) or the exonuclease activity of T4 DNA polymerase is used to generate complementary protruding 3′ termini. Because the overhangs created by the action of the exonucleases are slightly longer than the complementary sequence, after hybrid formation, a stretch of single-strand gap remains. This can be repaired in vivo (in the case of exonuclease III) or in vitro by the Klenow fragment of E. coli DNA polymerase I (in the case of T4 DNA polymerase).
Xi-Clone (Genlantis, Inc)
Xi-Clone, supplied by in kit form by its manufacturer, Genlantis, is used to clone PCR products directionally into expression vectors for high-level in vitro transcription and translation. An amplified segment of DNA is synthesized with single-stranded tails that are 28–32 nucleotides in length and homologous to the 5′ and 3′ ends of a linearized Xi-Clone plasmid. When the PCR fragments and the linear Xi-Clone vector are mixed and transformed into a recA strain of E. coli, the endogenous bacterial recombination system joins the two DNA fragments, generating a circular plasmid. Several linearized vectors are available from Genlatis, equipped, for example, for high-level expression of proteins in mammalian cells or in vitro.
The strengths of this system are its speed and its lack of reliance on in vitro enzymatically catalyzed reactions. Whether the system can be further developed to compete effectively with modular high-throughput recombination systems such as Gateway or In-Fusion is unclear.
Uracil DNA Glycosylase (UDG) Cloning
Using this method (Nisson et al. 1991; Rashtchian 1995), the primers used in PCR contain dUMP in their 5′-terminal regions. Digestion of the PCR products with UDG results in cleavage of the N-glycosidic bond between the deoxyribose residue and uracil. This cleavage generates abasic residues and destroys the base-pairing ability of the 5′-terminal sequences of the PCR products (Duncan 1981; Friedberg et al. 1981). Both the vector and the insert are amplified in reactions primed by uracil-containing primers and then treated with UDG. Thus, when the insert and vector are mixed, only one strand of insert is available for annealing with the complementary strand of the vector. Several linearized vectors are commercially available (Life Technologies) that carry defined 3′-protruding termini, 12 nucleotides in length. However, the method, although still used occasionally, never achieved great popularity and has largely been superseded by TOPO TA cloning and TOPO Tools (for a summary and comparison of these methods, see Table 3).
Ligase-free methods
TOPO TOOLS: CREATING LINEAR EXPRESSION CONSTRUCTS WITH FUNCTIONAL ELEMENTS
Chimeric DNA constructs remain the workhorses of much of molecular biology. Expression constructs typically consist of a series of functional units (promoters, open reading frames, signals for directing the traffic of protein to particular cellular locations, reporters, immunological tags, and terminators). Traditionally, these elements were joined together one at a time—often painfully slowly—by a combination of restriction enzyme digestion, ligation, and, when necessary, site-directed mutagenesis. The publication of the first paper outlining a PCR-based methodology for assembly and use of linear expression vectors (Sykes and Johnston 1999) opened the way to the development of functional DNA elements linked to vaccinia virus topoisomerase (TOPO Tools, Life Technologies; see Table 3). In this elegant system, the coding sequences of interest are first amplified using primers that introduce a vaccinia virus topoisomerase recognition site and ancillary sequences upstream and downstream of the coding sequence (see below). The A-tailed PCR products are then mixed with TOPO Tools elements, which are functional nucleic acid sequences (promoters, fusion tags, termination sequences, etc.) bound to topoisomerase, which catalyzes joining of the functional elements to the PCR products in a rapid (5–10 min) reaction at room temperature. After the desired 5′ or 3′ elements have been added, the resulting recombinants are amplified in a second PCR to generate functional linear cassettes that can be directly used in downstream applications (see Fig. 2). The technology can easily be adapted for use in 96-well or 393-well formats.
The manual on TOPO Tools Technology (Invitrogen 2002) contains clear, detailed descriptions of all of the steps and the necessary controls required to link user-generated PCR products to the 5′- and 3′-TOPO-adapted elements sold by Life Technologies. This information can be outlined as follows.
-
1. Design PCR primers to amplify the target sequence. These primers carry 5′-HO groups and should contain the 11-bp sequences required for joining the PCR product to TOPO Tools 5′ and 3′ elements.
-
2. Use a high-fidelity Taq polymerase (e.g., Platinum Taq, Life Technologies) to amplify the target sequence and, using gel electrophoresis, verify the integrity and concentration of the PCR product.
-
3. Mix the PCR product in the appropriate molar ratios with the 5′- or 3′-TOPO-adapted elements to create a linear construct.
-
4. Use element specific primers (supplied by Life Technologies) in a second round of PCR to amplify the target construct and its attached elements. Verify the integrity and concentration of the amplified product, which if everything has gone well, should be ready for transfection or other downstream applications.
Footnotes
-
From the Molecular Cloning collection, edited by Michael R. Green and Joseph Sambrook.








