
Allelic Exchange: Construction of an Unmarked In-Frame Deletion in Staphylococcus aureus
- 1Microbiology, School of Biological and Chemical Sciences, University of Galway, Galway H91 TK33, Ireland
- 2Center for Pandemic Vaccines and Therapeutics (ZEPAI), Paul-Ehrlich-Institute, 63225 Langen, Germany
- 3Section of Molecular Microbiology and Medical Research Council Centre for Molecular Bacteriology and Infection, Imperial College London, London, SW7 2AZ, United Kingdom
- ↵4Correspondence: a.grundling{at}imperial.ac.uk
Abstract
Here we describe an allelic-exchange procedure for the construction of an unmarked gene deletion in the bacterium Staphylococcus aureus. As a practical example, we outline the construction of a tagO gene deletion in S. aureus using the allelic-exchange plasmid pIMAY*. We first present the general principles of the allelic-exchange method, along with information on counterselectable markers. Furthermore, we summarize relevant cloning procedures, such as the splicing by overhang extension (SOE) polymerase chain reaction (PCR) and Gibson assembly methods, and we conclude by giving some general consideration to performing genetic modifications in S. aureus.
INTRODUCTION
The experimental strategies described herein were performed as part of a course in Advanced Bacterial Genetics delivered from 2017 to 2022. The overall aim of the methodologies presented here was to use the allelic-exchange procedure to construct an unmarked gene deletion in Staphylococcus aureus. The general principles of the allelic procedure will be outlined, and information on commonly used counterselectable markers aiding the procedure will be described. We also summarize how gene fragments can be fused (i.e., for the generation of in-frame gene deletions) by using the splicing by overhang extension (SOE) polymerase chain reaction (PCR) technique and how fragments can be cloned into a target vector using the Gibson assembly method. Finally, general considerations when performing genetic modifications in S. aureus will be discussed.
ALLELIC-EXCHANGE PROCEDURE
The ability to generate targeted gene deletions or point mutations in the chromosome is central to the investigation of the function of genes and their encoded products. Gene deletions or point mutations in Gram-positive bacteria are frequently constructed using an allelic-exchange procedure—this is an old, but reliable, and still widely used method. For the construction of gene deletions in S. aureus, the initial plasmid construction steps are performed in Escherichia coli using so-called “shuttle” vectors. These are plasmids that can be propagated both in E. coli and in S. aureus as they contain two origins of replication and either a selectable antibiotic-resistance marker that works in both organisms or one resistance marker for each host. Using the allelic-exchange method, the gene of interest can be replaced with a different gene, such as an antibiotic-resistance marker, a markerless in-frame deletion of the gene, or an otherwise modified gene (i.e., point mutations).
To replace a gene with an antibiotic-resistance marker, the expression of the resistance gene can be driven by the native promoter of the gene to be deleted. This, however, can be problematic if the gene is not constitutively expressed. Alternatively, a constitutive promoter can be introduced upstream of the resistance gene. This ensures that the selectable antibiotic marker is expressed—however, an associated problem is that this can also lead to inadvertent expression of downstream genes. In the strategies described here, a markerless in-frame gene deletion will be created. The advantage of a markerless in-frame deletion is that individual genes in large operons can be deleted while minimizing “polar effects” on the expression of downstream genes. Underpinning this strategy is allelic exchange, which makes use of the ability of bacteria to perform homologous recombination. To this end, DNA fragments of 500–1000 bp upstream of and downstream from the gene or operon to be deleted are amplified by PCR and fused by PCR or Gibson assembly and then inserted into a shuttle plasmid that can replicate normally in E. coli—but replicates only at low temperatures in S. aureus (Fig. 1A). This plasmid is first constructed in E. coli (see Protocol: Construction of a Staphylococcus aureus Gene-Deletion Allelic-Exchange Plasmid by Gibson Assembly and Recovery in Escherichia coli [Zeden et al. 2023a]) and subsequently introduced into S. aureus (see Protocol: Preparation of Electrocompetent Staphylococcus aureus Cells and Plasmid Transformation [Zeden et al. 2023b]). S. aureus colonies harboring the allelic-exchange plasmid are initially selected at a low temperature (usually 28°C–30°C) to enable plasmid replication. Next, the S. aureus colonies are grown at a high temperature (usually 37°C–43°C) while maintaining antibiotic selection. This enables growth of bacteria that have integrated the plasmid into the chromosome by a single crossover homologous-recombination event between the chromosomal DNA and the homologous plasmid-encoded DNA sequence (Fig. 1B). Homologous recombination can take place either in the upstream region (as shown in Fig. 1B) or downstream region. The result is the integration of the complete plasmid into the chromosome. Once the plasmid is integrated into the chromosome, the strains are grown at a temperature permissive for plasmid replication, but without antibiotic selection. Through a second recombination event, which can again take place either at the upstream or downstream homology region, the plasmid will excise from the chromosome. This can leave either the original wild-type copy of the gene in the chromosome or the desired gene deletion (Fig. 1C) (for the allelic-exchange methodology, see Protocol: Allelic-Exchange Procedure in Staphylococcus aureus [Zeden et al. 2023c]). PCR can be used to assess whether the wild-type copy of the gene or the desired gene deletion is present (see Protocol: Allelic-Exchange Procedure in Staphylococcus aureus [Zeden et al. 2023c]) or, for an S. aureus colony PCR method, see Protocol: Staphylococcus aureus Colony Polymerase Chain Reaction (Zeden et al. 2023d). Theoretically, there should be a 50% chance of obtaining a wild-type allele versus a gene deletion, but many factors can influence this percentage.
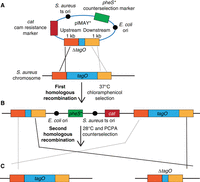
Schematic of an allelic-exchange procedure that uses plasmid pIMAY* and, by way of example, the generation of a tagO gene deletion. In A, the allelic-exchange plasmid with the gene deletion (ΔtagO) and 1 kb upstream and downstream gene fragments is shown along with the corresponding Staphylococcus aureus chromosomal tagO gene region. The plasmid has a temperature-sensitive origin (ts ori) of replication in S. aureus. (B) Growth at 37°C in the presence of antibiotic selection (chloramphenicol [cam] in this example) selects for strains that have undergone the first homologous-recombination event and now carry the plasmid integrated into the chromosome. (C) Following growth at 28°C and a second homologous-recombination event, strains either contain the wild-type gene copy or the desired gene deletion. The second homologous-recombination and plasmid-excision event is often selected for using a counterselectable marker. When using the allelic-exchange plasmid pIMAY*, the phenylalanine analog p-chlorophenylalanine (PCPA) can be used for counterselection, which is toxic for strains containing the plasmid.
COMMONLY USED COUNTERSELECTABLE MARKERS
The first homologous-recombination event is selected for by raising the temperature to one that is not permissive for plasmid replication, while maintaining the antibiotic selection. For the pIMAY* allelic-exchange plasmid described here, a temperature of 37°C is used. This temperature varies depending on the specific nature of the plasmid and its replication machinery and can be as high as 41°C–43°C. At this temperature, bacteria are selected in which the plasmid has integrated into the chromosome and hence no longer requires plasmid replication to be maintained. This first recombination step during the allelic-exchange procedure is usually very efficient. One problem that has been observed during this first step is the loss of the temperature sensitivity of the plasmid. Therefore, as outlined in Protocol: Allelic-Exchange Procedure in Staphylococcus aureus (Zeden et al. 2023c), it should be tested whether the allelic-exchange plasmid has retained the temperature-sensitive replication phenotype.
The rate-limiting step during the allelic-exchange procedure is often the second recombination event—the excision of the plasmid from the chromosome. However, one can select for the second homologous-recombination and plasmid-excision event using counterselectable markers. The allelic-exchange plasmid pIMAY* (Schuster et al. 2019), described herein, contains a mutated version of the pheS gene (Fig. 1A; for an additional map of the pIMAY* plasmid, see Fig. 6 in Protocol: Construction of a Staphylococcus aureus Gene-Deletion Allelic-Exchange Plasmid by Gibson Assembly and Recovery in Escherichia coli [Zeden et al. 2023a]). The gene pheS encodes the alpha subunit of the phenylalanine-tRNA synthetase, an enzyme required for the charging of tRNAs with phenylalanine. The PheS* variant contains a single amino acid substitution, leading to relaxed substrate specificity. When toxic phenylalanine analogs, such as p-chlorophenylalanine (PCPA), are supplied in the medium, bacteria expressing the PheS* variant are killed or show reduced growth compared with that of a strain only producing the wild-type PheS enzyme (Kast 1994). In the case of S. aureus and the pIMAY* plasmid, the addition of PCPA to the medium leads to reduced growth. Hence, by incorporating the pheS* gene into the allelic-exchange plasmid and supplying the toxic PCPA phenylalanine analog, one can screen for bacteria that have undergone the second homologous-recombination event and lost the allelic-exchange plasmid as these are not susceptible to PCPA.
There are several other counterselectable markers that are commonly used in bacteria (for review, see Reyrat et al. 1998). The original pIMAY plasmid frequently used for allelic exchange in S. aureus contains an antisense secY RNA as a counterselectable marker (Monk et al. 2012). In this case, part of the secY gene coding for SecY, an essential component of the protein-secretion translocon, is cloned in the antisense orientation under a tetracycline-inducible promoter. The addition of the nontoxic tetracycline analog anhydrotetracycline induces the expression of the antisense secY RNA, resulting in base-pairing with the secY mRNA and thereby preventing translation of SecY (Bae and Schneewind 2006; Monk et al. 2012). Other counterselectable markers frequently used are the tetAR genes that encode the tetracycline-resistance pump TetA and its regulator TetR, conferring tetracycline resistance to bacteria. Expression of the tetAR genes makes the cells not only resistant to tetracycline but also hypersensitive to lipophilic chelating agents such as fusaric acid and quinaldic acid. This enables selection of cells that have lost the tetAR genes by exposure to fusaric acid (Maloy and Nunn 1981).
Another widely used counterselectable marker is sacB. The sacB gene was isolated from Bacillus subtilis and encodes the enzyme levansucrase. Expression of sacB is harmless in most Gram-positive bacteria—but is lethal when expressed in Gram-negative bacteria in the presence of sucrose (Gay et al. 1985). The toxicity is believed to be due to the accumulation of high-molecular-weight fructose polymers called levans in the periplasm. The rpsL gene encodes the S12 protein component of the 30S ribosomal subunit. Certain mutations in the gene cause resistance to streptomycin. Resistance to streptomycin is recessive and hence the addition of a wild-type copy of the gene will lead to streptomycin sensitivity (Dean 1981). Therefore, the wild-type rpsL gene can be used as a counterselective marker in a strain that possesses also the mutant rpsL allele. Selection with streptomycin will only permit bacteria that have lost the wild-type rpsL gene to survive.
One other commonly used counterselectable marker is the ccdB gene, which encodes the toxin component of a toxin–antitoxin system located on the F-plasmid (Bernard et al. 1994). CcdB is a DNA gyrase inhibitor, which causes cell death in the absence of the antitoxin CcdA (Bernard et al. 1994). There is a known mutation in the gene encoding the DNA gyrase gene (gyrA462) that confers resistance to the toxin. This allows plasmids that contain ccdB to be propagated in a gyrA462 mutant strain without associated toxicity in the absence of ccdA. The ccdB gene is often inserted in cloning vectors and used to select against vectors lacking an insert, when transforming into a wild-type gyrA strain. The use of such plasmids can very efficiently eliminate the recovery of transformants containing the unmodified vector. However, one needs to keep in mind that ccdB does not function as a counterselectable marker in some commonly used cloning strains such as E. coli XL1-Blue, which contains an F-plasmid and hence expresses the antitoxin CcdA.
SOE PCR
One method that is commonly used to produce the required DNA fragments for the construction of gene deletions is SOE PCR (Horton et al. 1989). In our example here, an S. aureus tagO gene-deletion fragment is created. For this, the first 30 bases are fused to the last 30 bases of the tagO gene and flanked by ∼1-kb chromosomal DNA fragments upstream of and downstream from the resulting tagO gene deletion. Such a fragment can be generated using four appropriately designed primers and two consecutive rounds of PCRs (Fig. 2). For the first round, two PCRs in separate tubes are set up. Primers P1 and P2 are used to amplify the upstream 1-kb homology regions (red) and the first 30 bases of tagO (dark blue). The forward primer P1 also contains restriction-site sequences or sequences homologous to the plasmid DNA (indicated in black) for the subsequent Gibson cloning step (Fig. 2). Primers P3 and P4 are used to amplify the last 30 bases of the tagO gene and the downstream 1-kb homology region. As described for primer P1, primer P4 contains sequences recognized by a specific restriction enzyme or sequences homologous to the plasmid DNA for the subsequent cloning step. To be able to fuse the two PCR products in the second round of PCR, the internal primers P2 and P3 contain complementary 5′ extensions. In the case of primer P3, this extension is complementary to the front part of the tagO gene (dark blue extension), and, in the case of primer P2, this extension is complementary to the back part of tagO (green extension). Usually, 12–15 bases are added to the 5′ end of these primers, which leads to an overlapping region of ∼24–30 bases. Once the two PCR fragments of the upstream and downstream regions have been produced, they are gel-purified to remove any excess primer. They are then used as template DNA in a second PCR using the outside primers P1 and P4, leading to the generation of a fusion product. In this manner, any two pieces of DNA can be fused together at any point without leaving a “scar.”
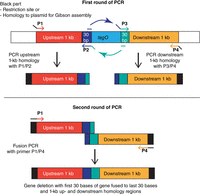
Schematic representation of the splicing by overhang extension (SOE) polymerase chain reaction (PCR) method. Here, as an example, the primer design for the construction of a tagO gene deletion fragment is shown, in which the first 30 bases of the gene are fused to the last 30 bases of the gene, which is flanked by 1-kb homology regions at the front and rear of the gene.
GIBSON ASSEMBLY METHOD
In our associated protocols, the Gibson assembly method is used to insert the upstream and downstream 1-kb DNA fragments flanking the tagO gene into the allelic-exchange shuttle plasmid pIMAY*. This method uses an “isothermal, single-reaction method for assembling multiple overlapping DNA molecules by the concerted action of three enzymes” (Gibson et al. 2009). These enzymes are a 5′ exonuclease, a DNA polymerase, and a DNA ligase. The principle regarding how DNA fragments with overlapping sequences can be fused in a Gibson assembly reaction is shown in Figure 3. First, the 5′ exonuclease chews back one of the strands of the DNA fragments, exposing single-stranded homologous regions that can anneal to each other. Next, the DNA polymerase fills in any gaps that are present after the strands have annealed. Finally, the ligase joins the nicks in the sequence to make a single fragment (Fig. 3).
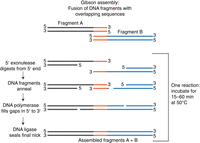
Principle of the Gibson assembly method permitting the joining of DNA fragments possessing overlapping sequences through the concerted action of three enzymes. Figure adapted from information found on the New England BioLabs (NEB) website describing the Gibson Assembly Cloning Kit (NEB E5510).
For the construction of the gene-deletion allelic-exchange plasmid, the first step is the amplification of the upstream and downstream DNA fragments and the addition of vector-specific sequences to one end of the fragment and overlapping complementary regions to the other end of the fragment. These sequences are introduced by appropriately designed primers used during the PCR amplification step (Fig. 4). The primers must have two sequence components—an overlap region for the Gibson assembly reaction and the gene-specific sequence, required for priming during the PCR amplification. The overlap regions should be between 15 and 25 bp (but can also be longer) and should have an annealing temperature of 48°C or higher. Primers can be designed manually, as described above for the SOE PCR method, but the primers will need their annealing temperatures checked carefully. Alternatively, a NEBuilder Assembly tool is available on the NEB website for the primer design (nebuilder.neb.com). The plasmid is linearized with restriction enzymes to expose the homologous regions used for integration at the ends. The PCR product and the cut vector are then incubated for 15–60 min at 50°C together with a 5′ exonuclease, DNA polymerase, and DNA ligase to fuse the two PCR fragments and the vector to generate the gene-deletion allelic-exchange plasmid. This plasmid is then introduced into E. coli by transformation.
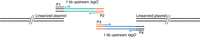
Schematic representation of the generation of the gene-specific allelic-exchange plasmid using the Gibson assembly method. The 1-kb upstream and downstream gene deletion fragments are generated by polymerase chain reaction (PCR). Overlapping sequences to the plasmid vector (black regions) and for fusing the two gene fragments (orange regions) are introduced as part of the primer sequences.
WORKING WITH STAPHYLOCOCCUS AUREUS
To create mutations in S. aureus, the first cloning steps are performed in standard E. coli cloning strains, and the plasmids are then subsequently introduced by electroporation into S. aureus. In the experimental methodologies described here, we typically use the S. aureus strain RN4220 (Kreiswirth et al. 1983). RN4220 is a commonly used restriction-deficient mutant laboratory strain that can be readily transformed with DNA isolated from E. coli (Kreiswirth et al. 1983). Its restriction deficiency is due to the inactivation of HdrR, a restriction enzyme that is part of a type I DNA restriction modification (RM) system (Waldron and Lindsay 2006). Different S. aureus clonal complex (CC) strains exist, and they express different type I RM systems and hence recognize and methylate different DNA sequences. Strain RN4220 is part of the clonal complex 8 (CC8). Because of nonspecific mutagenesis, several mutations have been introduced into the RN4220 genome (Nair et al. 2011; Berscheid et al. 2012; Bæk et al. 2013), and hence this strain is usually only used as a cloning intermediate strain and no longer for physiological or virulence studies. As strain RN4220 is only restriction deficient but can still modify DNA, plasmids isolated from this strain can subsequently be efficiently introduced into other CC8 S. aureus strains, such as the clinically relevant methicillin-resistant S. aureus (MRSA) USA300 strains (i.e., strains LAC or LAC*). More recently, E. coli strains expressing the type I RM system from S. aureus have been produced (Monk et al. 2015). One such strain is E. coli strain IM08B (Monk et al. 2015). DNA isolated from this strain can be readily transformed not only into S. aureus RN4220 but also into clinically relevant CC8 S. aureus strains. Hence, the use of E. coli strain IM08B bypasses the need for passing plasmids through S. aureus strain RN4220. The majority of clinical S. aureus isolates also contain an active type IV restriction system, which cleaves DNA that contains methylated cytosines (Xu et al. 2011). DNA in E. coli is usually methylated by the Dcm methyltransferase, which methylates the second cytosine in the sequences CCAGG and CCTGG. Hence, in addition to expressing the S. aureus CC8 type I RM system, E. coli strain IM08B is also a dcm− mutant strain that avoids the degradation of the DNA by the type IV restriction system when introduced into S. aureus. Similar types of RM systems might be active in other bacterial species and need to be considered when setting up genetic systems and attempting to perform genetic manipulations in bacteria other than well-characterized model organisms.
OVERVIEW OF THE EXPERIMENTAL METHODOLOGIES
Step-by-step guidance for the construction of an in-frame gene deletion in S. aureus using the allelic-exchange procedure and the final confirmation of the gene deletion by PCR is presented in four protocols associated with this overview. By following these protocols, marker-less gene deletions can be generated in S. aureus using the allelic-exchange method. To make the practical aspects of the primer design and confirmation of the gene deletion easier to follow, we present a specific example for the construction of a tagO mutant in the S. aureus strain RN4220. To this end, we show how the Gibson assembly method can be used to fuse the first 30 and last 30 bases of tagO and clone ∼1-kb upstream and downstream DNA regions of the tagO gene into the allelic-exchange plasmid pIMAY*. This is followed first by recovery of the resulting plasmid pIMAY*-ΔtagO in E. coli and then confirmation of the veracity of the insert by colony PCR and sequencing (see Protocol: Construction of a Staphylococcus aureus Gene-Deletion Allelic-Exchange Plasmid by Gibson Assembly and Recovery in Escherichia coli [Zeden et al. 2023a]). Next, we detail the method for preparing electrocompetent cells of S. aureus strain RN4220 and then introducing the plasmid pIMAY*-ΔtagO into these cells by electroporation. Colonies containing the plasmid can then be selected on chloramphenicol plates at a low temperature (28°C), which is permissive for plasmid replication (Protocol: Preparation of Electrocompetent Staphylococcus aureus Cells and Plasmid Transformation [Zeden et al. 2023b]). Thereafter, we describe how the allelic-exchange procedure can be performed in S. aureus. Bacteria are grown in the presence of antibiotic selection (chloramphenicol) at a high temperature (37°C), which is not permissive for plasmid replication. This will select for bacteria in which the plasmid has integrated into the chromosome by homologous recombination. Subsequently, the cultures are shifted back to 28°C and passaged for several generations in the absence of antibiotic selection to allow for plasmid excision. Finally, S. aureus strains that have lost the plasmid and undergone the second recombination event are selected/screened by using a counterselection procedure on plates containing PCPA. The resulting strains, which have lost the plasmid, are antibiotic-sensitive and will contain either an intact wild-type copy of the gene or the desired gene deletion. Genomic DNA is then isolated from the resulting antibiotic-sensitive clones, and the identity of the gene region of interest can then be assessed by PCR (see Protocol: Allelic-Exchange Procedure in Staphylococcus aureus [Zeden et al. 2023c]). Finally, we give details of a more rapid S. aureus colony PCR method for assessing whether the resulting chloramphenicol-sensitive S. aureus colonies have a wild-type gene copy or the desired gene deletion (Protocol: Staphylococcus aureus Colony Polymerase Chain Reaction [Zeden et al. 2023d]).
ACKNOWLEDGMENTS
The research in the Gründling laboratory is supported by the Wellcome Trust grant 210671/Z/18/Z/WT. M.S.Z. is supported by the Health Research Board grant ILP-POR-2019-102.
Footnotes
-
From the Experiments in Bacterial Genetics collection, edited by Lionello Bossi, Andrew Camilli, and Angelika Gründling.








