
Use of Phage Display and Other Molecular Display Methods for the Development of Monobodies
- Akiko Koide1,2 and
- Shohei Koide1,3,4
- 1Laura and Isaac Perlmutter Cancer Center, New York University Langone Health, New York, New York 10016, USA
- 2Department of Medicine, New York University School of Medicine, New York, New York 10016, USA
- 3Department of Biochemistry and Molecular Pharmacology, New York University School of Medicine, New York, New York 10016, USA
- ↵4Correspondence: shohei.koide{at}nyulangone.org
Abstract
Synthetic binding proteins are human-made binding proteins that use non-antibody proteins as the starting scaffold. Molecular display technologies, such as phage display, enable the construction of large combinatorial libraries and their efficient sorting and, thus, are crucial for the development of synthetic binding proteins. Monobodies are the founding system of a set of synthetic binding proteins based on the fibronectin type III (FN3) domain. Since the original report in 1998, the monobody and related FN3-based systems have steadily been refined, and current methods are capable of rapidly generating potent and selective binding molecules to even challenging targets. The FN3 domain is small (∼90 amino acids) and autonomous and is structurally similar to the conventional immunoglobulin (Ig) domain. Unlike the Ig domain, however, the FN3 lacks a disulfide bond but is highly stable. These attributes of FN3 present unique opportunities and challenges in the design of phage and other display systems, combinatorial libraries, and library sorting strategies. This article reviews key technological innovations in the establishment of our monobody development pipeline, with an emphasis on phage display methodology. These give insights into the molecular mechanisms underlying molecular display technologies and protein–protein interactions, which should be broadly applicable to diverse systems intended for generating high-performance binding proteins.
INTRODUCTION
Developing custom reagents that selectively and potently bind a target molecule of interest is a core biotechnological methodology with broad utility. The ability of the adaptive immune system to generate antibodies that can bind diverse antigens after animal immunization has been exploited for decades for the development of research reagents and therapeutics, and analysis of the immune system and antibody–antigen interactions has provided key insights into the underlying molecular mechanisms. The key aspects of this process involve the generation of molecular diversity (Tonegawa 1983) and the selection of antibody clones of interest (Burnet 1976; Hodgkin et al. 2007). Each B cell expresses a single antibody on its surface, which establishes the linkage between the sequence of an antibody (i.e., genotype) and its function (i.e., phenotype) (Nossal and Lederberg 1958; Melchers and Andersson 1974). The invention of phage display made it possible to effectively apply this principle of genotype–phenotype linkage at the level of protein function outside the context of the immune system (Smith 1985).
The ability of the natural immune system and early successes of phage display in generating functional antibody fragments and short peptides have inspired the genesis of the field of synthetic binding proteins (Skerra 2000; Binz et al. 2005; Koide 2010). Similar to how natural antibodies with different binding specificity are generated primarily with different complementarity-determining regions that collectively form a contiguous surface of the otherwise mostly invariant immunoglobulin molecule (Mariuzza et al. 1987), synthetic binding proteins are generated by altering portions of a functionally inert protein, referred to as a protein scaffold. One might envision that, by starting with a well-chosen scaffold, one can achieve high binding function with additional desirable properties, such as small size, high stability, ease of production, and ease of use as a building block for constructing multidomain, multifunction proteins. A common goal of developing a synthetic binding protein system is to have the ability to generate proteins that bind to diverse target molecules, similar to that of the natural immune system.
Over the years, our community has collectively solved the challenge of generating highly functional synthetic binding proteins. There are now a number of well-established systems, including anticalin (Richter et al. 2014), affibody (Feldwisch and Tolmachev 2012), DARPin (Plückthun 2015), and monobody (Hantschel et al. 2020). In this article, we provide a historical account of the development of the monobody system, with particular emphasis on illustrating technological challenges and breakthroughs, which help us understand the molecular mechanisms underlying phage display and protein–protein interaction and should be applicable to diverse synthetic binding protein systems.
The Monobody System
Monobody is a synthetic binding protein that is built from the molecular scaffold of the fibronectin type III (FN3) domain. The use of FN3 as a scaffold was first reported in 1998 by our group (Koide et al. 1998). Our vision was that, whereas immunoglobulins are effective scaffolds for creating binding proteins, that is, antibodies, it may be possible to establish a simpler system for generating binding proteins against a protein of interest. At the time of the inception of the monobody system, FN3 appeared to be a particularly attractive scaffold, as described below.
FN3 is a small, autonomously folded domain with a β-sandwich architecture (Fig. 1). It is among the most commonly occurring domains in mammalian proteomes, present in both extracellular and intracellular proteins. Indeed, according to the SMART database, the human proteome has 4104 FN3 domains in 673 proteins (smart.embl-heidelberg.de/smart/do_annotation.pl?DOMAIN=FN3). FN3 is a member of the immunoglobulin (Ig) superfold, with seven β-strands, and its β-strand topology is similar to that of the Ig variable domain, although FN3 has only seven β-strands (Fig. 1A). A key difference between FN3 and the conventional Ig domain is that FN3 lacks an intradomain disulfide bond that characterizes conventional Ig domains (Fig. 1A). Nevertheless, many FN3 domains have high conformational stability (Plaxco et al. 1996; Koide et al. 1998; Cota et al. 2000; Hamill et al. 2000; Jacobs et al. 2012). Therefore, we originally anticipated that it would be easier to produce FN3-based binding proteins, for example, by cytoplasmic overexpression in Escherichia coli. This is because the reducing environment of the cytoplasm makes it challenging to produce conventional Ig molecules, as their folding usually depends on the formation of disulfide bonds. Indeed, we and others have shown that monobodies can be expressed at a high level in their functional form in the cytoplasm of E. coli as well as in eukaryotic cells (Koide et al. 1998, 2002; Wojcik et al. 2010; Grebien et al. 2011).
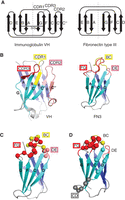
Comparison of immunoglobulin heavy-chain variable (VH) and fibronectin type III (FN3) domains and monobody library designs. (A) β-Strand and loop topology. The two β-sheets of the corresponding domains are shown in an open book manner with the dashed lines indicating the boundary between the two β-sheets. SS denotes an intradomain disulfide bond. (B) Cartoon representation of VH (PDB ID: 1VFB) and FN3 (3CSB). The complementarity-determining regions (CDRs) are according to the Kabat definition (Wu et al. 1993). (A,B, Drawn based on Fig. 1 in Koide et al. 1998.) (C,D) Two distinct types of monobody library designs. An antibody-like library design is depicted in C, and a non-antibody-like, “side” library is shown in D. (C,D, Modified from Fig. 1 in Koide et al. 2012a, with permission.)
Despite the many attractive attributes of FN3, it is not used by the adaptive immune system. Therefore, there were no immediate sources of sequence diversity equivalent to the natural repertoires of B-cell receptors (including antibodies) and T-cell receptors. Consequently, key innovations in the history of monobody development included the establishment of the phenotype–genotype linkage using molecular display systems and the design of effective combinatorial libraries (Koide et al. 1998, 2012a; Wojcik et al. 2010).
One could, in principle, use any FN3 domain from the thousands available in databases to build a new FN3-based system. In reality, however, only a small number of natural and designed FN3 molecules have been adopted as molecular scaffolds for developing synthetic binding proteins. In addition to the tenth FN3 of human fibronectin used for monobodies and adnectins (Xu et al. 2002), a consensus FN3 domain and one from human tenascin have been used for constructing the centyrin and TN3 systems, respectively (Jacobs et al. 2012; Oganesyan et al. 2013). This limited variety is not surprising, because it takes substantial effort to establish an effective system for generating binding proteins even when using a single scaffold, as we discuss below for the monobody system. Furthermore, now that there already are well-established systems, including monobody, DARPin, anticalin, and affibody (Sha et al. 2017), it is our opinion that the field has reached the point of diminishing returns in terms of developing a synthetic binding protein system using a novel scaffold.
Phage Display of Monobodies
At the time of the inception of the monobody concept, that is, the use of FN3 as a scaffold for the development of synthetic binding proteins, phage display was an obvious choice as a molecular display platform for constructing libraries and performing library sorting. We started with the then-standard system that fused the FN3 scaffold to a carboxy-terminal fragment of p3 and p8 of the M13 phage (Koide et al. 1998; Sidhu et al. 2000; Richards et al. 2003). This fusion gene was placed under the control of the lac promoter (Koide et al. 1998). This system was considered sufficiently effective for generating the first set of monobodies, which showed what was considered at the time to be moderate affinity. In retrospect, however, the affinity and selectivity of the monobodies from early studies are nowhere near those that we can achieve using current technologies (Sha et al. 2017; Hantschel et al. 2020; Akkapeddi et al. 2021). Subsequent studies identified challenges in phage display associated with high stability and rapid folding of FN3, and developed strategies to overcome them, as discussed below.
For effective recovery of functional clones, that is, clones that bind to a target of interest, it is crucial that all clones encoded by a phage display library are displayed on the phage surface, with minimal bias. We noticed low levels of surface display of monobodies using our original vector, which likely limited our ability to identify functional clones. Phage display systems based on M13 or fd phage require that the protein to be displayed is secreted, along with the extracellular portion of a phage coat protein, that is, p3 and p8, into the periplasm of host E. coli (Petrenko and Smith 2005). Such secretion is enabled by the attachment of a signal sequence to the amino terminus of the protein of interest. The early generations of phage display vectors used signal sequences that mediate Sec-dependent, posttranslational secretion, which requires the fully translated protein to be unfolded as it is translocated across the membrane. These vectors were developed for linear peptides, disulfide-constrained peptides, and antibody fragments, which are either natively disordered or largely disordered until disulfide bonds are formed in the oxidizing environment of the extracellular milieu and the lumen of the secretion pathway, that is, the ER and Golgi. In other words, these molecules are disordered until they have been transferred across the plasma membrane, and are unlikely to obstruct the translocation process. In contrast, FN3 is highly stable and rapidly folds into its native globular conformation (Plaxco et al. 1996), and it is likely to present a substantial energetic barrier for the translocation process.
An important innovation came from Steiner et al. (2006), who showed that replacing a Sec-dependent signal sequence with a signal recognition particle (SRP)-dependent sequence dramatically increases the surface display level of another highly stable, rapidly folding protein, DARPin. We found that replacing a Sec-dependent signal sequence with an SRP-dependent signal sequence from E. coli DsbA similarly increases the display level of the monobody-p3 fusion on M13 phage by 100-fold (Wojcik et al. 2010). We also found that one can further increase the display level by the use of a mutant helper phage called hyperphage (Rondot et al. 2001) in conjunction with fusing the monobody to the full-length p3 instead of only to the carboxy-terminal half of p3, which is required for making the resulting phage particles infectious.
Using this “SRP phage display” system, we systematically examined conditions that potentially affect the display level. As expected, the number of phage particles in the culture supernatant depended nonlinearly on the length of culture and, consequently, it is important to experimentally determine an appropriate duration of phage propagation. Interestingly, the aeration of the E. coli culture for propagating phages strongly affected the display level. Phage particles produced with a non-baffled flask and slow shaking, that is, shaking just sufficient to maintain cell suspension, showed up to 50 times higher levels of monobody display than those produced using a baffled flask and vigorous shaking (Wojcik et al. 2010). In contrast, we did not observe such dependence on aeration of the display level of a Fab phage vector. Although we have not elucidated the molecular mechanism underlying this dependence of the display level on culture conditions, these results further underscore that phage display of a highly stable protein imposes stress on the host E. coli cells and that culture conditions suitable for rapid E. coli growth do not necessarily produce phage particles suitable for effective library sorting.
Monobody Library Designs
The most common approach to generating synthetic binding proteins is to design a combinatorial library in which amino acid diversity is introduced at positions of a scaffold in such a way that the diversified positions are expected to form a contiguous surface for interacting with a target of interest. The absence of a natural, immune-like repertoire for FN3 presents both challenges and opportunities in combinatorial library design. The amino acid diversity needs to provide new interactions and also minimize negative impacts on biophysical properties such as stability. The size of a molecular display library that can be experimentally interrogated in a meaningful manner (up to approximately 1013 depending on display method) is much smaller than the total number of possible sequences that can be encoded even in a small library design, for example, 3 × 1019 for a total of 15 fully randomized positions using one codon for each amino acid. Consequently, it is unlikely that the positions and amino acid diversity at each position can be optimized by an exhaustive search of fully randomized libraries.
There are two important factors for designing libraries: positions to be diversified and chemical diversity (which amino acid residues to be used and their ratio). Given the structural homology of FN3 with immunoglobulin G (IgG), our initial approach was to introduce diversity in the loops that are structurally equivalent to the complementarity-determining regions (CDRs) of immunoglobulins (Koide et al. 1998). However, it was unclear which loops of FN3 can be extensively modified without a drastic reduction in stability. For example, one poorly chosen mutation could denature the protein and make it nonfunctional and, thus, such a mutation should be avoided in library designs. We systematically tested the effects of altering loop regions of the monobody scaffold by insertion mutagenesis, which revealed that loops except for the EF loop can tolerate mutations (Batori et al. 2002). The EF loop has a feature called tyrosine corner that is important for the structural integrity of FN3 (Hamill et al. 2000). Therefore, we have kept the EF loop as wild-type in our libraries. This restriction makes it difficult to construct a library with diversification in multiple loops using the “bottom” end of the molecule (Koide et al. 2002). In parallel, we identified a stabilizing mutation at a site distal to the loops (Fig. 1B) (Koide et al. 2001). These biophysical studies provided foundational knowledge for designing libraries.
Three loops located at the “top” part of the FN3 protein, BC, DE, and FG (Fig. 1B), tolerate extensive mutations (Batori et al. 2002) and have thus been used as sites for constructing combinatorial libraries (Fig. 1C). Monobodies are stable even after mutation at more than 20% of the amino acid residues (Parker et al. 2005; Koide et al. 2012a).
Early libraries using diversification schemes with the NNK codons that encode all 20 amino acids, produced only low-affinity monobodies (Koide et al. 1998, 2002), which prompted us to examine the importance of chemical diversity. An important breakthrough was the finding that a monobody library using a binary code consisting of only Tyr and Ser but with varied loop lengths, produced high-affinity monobodies with good specificity (Koide et al. 2007). This work was inspired by pioneering studies by the Sidhu group that established the effectiveness of “reduced codes,” including the Tyr/Ser binary code, in the Fab scaffold (Fellouse et al. 2004, 2005). The effectiveness of the Tyr/Ser binary code in the much smaller monobody scaffold was still surprising. Structural studies of these molecules defined the dominant roles of Tyr in making contacts in the interface (Koide and Sidhu 2009). Because the size of a combinatorial library using a binary code (e.g., 220 = ∼1 million) is small enough to be fully sampled in a phage display library, studies using such libraries determine the effectiveness of library designs, as they are not affected by limited, stochastic sampling of encoded sequences that plague libraries that use more expanded codes.
The simplicity of the binary library enabled us to examine the effects of expanding the code. As one might expect, including additional amino acids at lower frequencies than Tyr and Ser produced more functional monobodies (Gilbreth et al. 2008). Structural studies revealed that Tyr still plays the dominant role in the interface of a monobody with an expanded code, and that the additional amino acid types mainly contribute to improving the shape complementarity of the interface (Gilbreth et al. 2008; Gilbreth and Koide 2012). An independent study confirmed the utility of additional amino acid types (Hackel and Wittrup 2010). Subsequent iterations including parallel studies of Fab libraries (Fellouse et al. 2007; Wojcik et al. 2010; Miller et al. 2012) have resulted in a design that has proven to be highly effective in producing monobodies for diverse targets. Such a design uses the following composition: 30% Tyr, 15% Ser, 10% Gly, 5% Phe, 5% Trp, and 2.5% each of all the other amino acids except for Cys (Koide et al. 2012a). Subsequently, others have developed similar “antibody-like” libraries (Xu et al. 2002; Olson and Roberts 2007; Hackel et al. 2008; Wensel et al. 2017; Lipovšek et al. 2018; Kondo et al. 2020).
Despite these advances, we found a unique challenge in the production of phage display libraries of monobodies. Large combinatorial libraries in the phage display format are often constructed using Kunkel mutagenesis (Kunkel 1985; Kunkel et al. 1987) and electroporation of the SS320 E. coli strain as the host (Sidhu et al. 2000). Because the efficiency of Kunkel mutagenesis is typically 50%–80%, a library constructed with this method contains a substantial fraction of the template construct used for mutagenesis. A construct that contains a termination codon, which is eliminated upon mutagenesis, for example, in a CDR, is often used as the template. In this manner, the “stop template,” which does not display the protein of interest on the phage, is depleted in a library sorting process. In the case of phage display of monobodies, phage particles containing a stop template propagated more rapidly than monobody-displaying phage particles during the amplification step, dominating the amplified library and overriding the enrichment of target-binding clones. This growth bias is probably due to the stress of producing monobody-p3 fusion proteins on E. coli. As a workaround, we used a template construct that does display a monobody clone but it can be selectively removed by restriction enzyme digestion of purified DNA.
The use of synthetic libraries rather than natural immune libraries offers complete freedom in library design. Whereas antibody-like libraries have produced highly functional monobodies, structural studies of these monobodies in complex with their respective targets have revealed mismatches between the library design and the actual surfaces of monobodies used for contacting their targets. A subset of monobodies use the FG loop and β-strand regions of the scaffold for making contacts with their targets, rather than the three diversified loops (Gilbreth and Koide 2012; Koide et al. 2012a). These observations inspired us to design a distinct library in which we diversify residues in the CD and FG loops that are located on the opposite ends of the monobody molecule (Fig. 1D) and in the strands that connect these loops. This “side” library shows distinct interface topography from that of the antibody-like, “loop” libraries and, thus, is capable of generating monobodies that bind to flatter surfaces in target molecules (Koide et al. 2012a). In retrospect, it is not surprising that we have been able to develop monobodies that use this surface for target recognition, because it corresponds approximately to the heterodimerization interfaces in immunoglobulin G (IgG), one between the heavy-chain variable (VH) and light-chain variable (VL) domains and one between the heavy-chain constant 1 (CH1) and light-chain constant (CL) domains, as well as the homodimerization interface of the heavy-chain constant 3 (CH3) domain, which are all high-affinity interactions. In the conventional IgG molecules, this surface is already taken, and not available for antigen recognition. Other groups have followed our design and developed similar libraries (Diem et al. 2014; Wensel et al. 2017; Chan et al. 2019). Having two distinct types of libraries greatly improves the likelihood of developing highly functional monobodies to diverse targets.
Other Display Systems for Monobody Discovery
The robustness of FN3 proteins has made monobody and other FN3 scaffolds compatible with virtually all molecular display systems. In addition to phage display, mRNA display (Xu et al. 2002; Olson and Roberts 2007; Kondo et al. 2020), yeast two hybrid (Koide et al. 2002), DNA display (Diem et al. 2014), and yeast display (Hackel et al. 2008; Koide et al. 2012a) have all been used.
Yeast display, for instance, has been used for iterative improvement of affinity, which have yielded molecules with low pM KD values (Hackel et al. 2008). Yeast display has also been used as the secondary screening method for clones enriched from larger primary libraries constructed in the phage and mRNA display formats (Koide et al. 2012a,b). The combination of phage/mRNA and yeast display complements their strengths and minimizes their weaknesses, such as the difficulty in quantitative sorting with phage/mRNA display and the difficulty in constructing large libraries with yeast display. We find the combination of phage display and yeast display to be particularly effective in comprehensively characterizing an enriched pool from sorting of a phage display library and in discovering clones with exquisite selectivity. In phage display, enriched clones, typically dozens to hundreds, are individually produced and screened using phage ELISA. This process may miss rare clones with desirable properties. Yeast display coupled with a fluorescence-activated cell sorter can screen millions of cells and, thus, can interrogate the entire pool of clones in an enriched phage-display library. Furthermore, it is straightforward to implement both positive and negative sorting in yeast display. We have successfully developed monobodies with exquisite specificity and high potency using this approach (Akkapeddi et al. 2021; Teng et al. 2022).
CONCLUSIONS
Phage display and other molecular display technologies continue to play central roles in the discovery of monobodies and other FN3-based synthetic binding proteins. Numerous molecules have been reported for diverse biomedical applications, including therapeutics that have successfully completed phase II clinical trials (NCT02515669, NCT03549260; see also Stein et al. 2019) and over 75 crystal structures of monobody-target complexes available in the Protein Data Bank (Fig. 2). Describing specific details of these individual molecules is beyond the scope of this article. The reader, however, is referred to recent reviews for a discussion on monobodies for therapeutic and diagnostic applications (Chandler and Buckle 2020), intracellular target discovery and validation (Akkapeddi et al. 2021), and structural and mechanistic investigation (Hantschel et al. 2020). The expanding efforts and innovations made by the community will define applications that exploit unique strengths of the monobody molecules, including small size, high stability and rapid folding, the absence of disulfide bonds, and the ease of constructing fusion proteins (Fulcher et al. 2017; Donnelly et al. 2018; Kulemzin et al. 2018; Ludwicki et al. 2019; Röth et al. 2020; Lim et al. 2021; Robu et al. 2021). These successes in developing novel synthetic binding proteins may now appear routine, but they are the culmination of multidisciplinary research. The ambition of establishing a robust system that consistently generates potent and selective binding proteins to diverse targets has challenged the protein engineering community both intellectually and technologically. Indeed, to establish such a system, one needs to become adept at display technologies, protein–protein interactions, protein stability and folding, and microbiology. Practitioners of display technologies and synthetic binding protein development would be well served by acquiring a deep understanding of the mechanistic underpinnings of these powerful technologies.
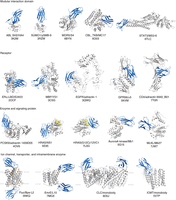
Examples of crystal structures of monobodies and adnectins in complex with their respective target proteins. The examples are divided based on the type of target proteins. Monobodies and adnectins are shown in blue and the targets in gray, with the epitope in orange. For HRAS, the bound nucleotide is show in yellow. The target name, binder name, and Protein Data Bank (PDB) ID are shown for each structure. (ABL) Abelson tyrosine-protein kinase, (SH2) src homology 2, (SUMO) small ubiquitin-related modifier, (WDR5) WD repeat-containing protein 5, (CBL) E3 ubiquitin-protein ligase CBL, (TKB) tyrosine kinase binding, (STAT3) signal transducer and activator of transcription 3, (ERα) estrogen receptor α, (LBD) ligand-binding domain, (MBP) maltose-binding protein, (EGFR) epidermal growth factor receptor, (GRP56) G-protein coupled receptor 56, (PCSK9) proprotein convertase subtilisin/kexin type 9, (MLKL) mixed-lineage kinase domain-like protein, (Fluc) fluoride channel, (CLC) chloride channel, (ICMT) protein-S-isoprenylcysteine O-methyltransferase.
Footnotes
-
From the Advances in Phage Display collection, edited by Gregg J. Silverman, Christoph Rader, and Sachdev S. Sidhu.








