
Principles of Affinity Selection
- ↵1Correspondence: smithgp{at}missouri.edu
Abstract
The most common application of phage-display technology is the discovery of peptides or proteins that specifically bind some molecule or other substance of interest—for example, antibodies that specifically bind an antigen. The discovery process starts with a library encompassing a very large array of proteins or peptides with a great diversity of binding specificities—for example, single-chain antibodies with a great diversity of antigen-binding sites. Each member of the array is displayed on the surface of hundreds to billions of identical virus particles (virions) belonging to a single-phage clone; the library as a whole comprises millions to billions of such clones, all mixed together in a single vessel. Affinity selection is the process by which a molecule or substance of interest—generically called the selector—is used to select very rare clones in the library displaying proteins or peptides that happen to bind the selector with high affinity and selectivity. Here, I explain general principles guiding a successful affinity-selection project—principles grounded in phage biology, kinetics of reversible binding, technological advances, and the practical experience of thousands of investigators around the globe.
BACKGROUND
Here I summarize fundamental principles to consider in designing an affinity-selection project: that is, a project with the goal of discovering peptides or proteins that bind specifically to some chosen molecule or other substance, which is called the selector. Affinity selection is often called “panning” in laboratory jargon, reflecting the phage display's origin in immunology (Wysocki and Sato 1978), but that is much too narrow a term to cover the common realizations of the selection process. The principles explained here are grounded in basic reversible binding kinetics, phage biology, technological advances, and practical experience.
There are numerous realizations of the general phage-display concept, involving different phages or phage proteins; most of the principles apply to all of them. But here the principles are shown specifically for systems in which the peptides or proteins are displayed on coat protein III (pIII) of the Ff family of filamentous phages: derivatives of wild-type strains fd, f1, and M13 (Rakonjac et al. 2017). Even within that category, innumerable innovative variations have been investigated by the global phage-display community. Here, however, only the most common, “mainstream” procedures are covered explicitly.
THE AFFINITY-SELECTION PROCESS
The upper part of Figure 1 shows a side-view cartoon of the long, thin phage particle—called the virion—showing the basic structural features involved in the affinity-selection process (Rakonjac et al. 2017). There is a coiled, single-stranded, circular chromosome inside the particle, represented by a black chain with colored segments. The colored segments correspond to one of the chromosome's genes: a recombinant form of phage gene III, encoding pIII. The parts of the recombinant gene that derive from the wild-type gene III are represented by yellow segments, whereas the foreign (i.e., nonphage) insert that encodes the displayed peptide is represented by a pink segment. The shorter yellow segment encodes the amino-terminal signal peptide of pIII, which is removed in the process of virion assembly. The pink segment plus the longer yellow segment encode the mature form of the recombinant pIII in the completed virion, with the displayed foreign peptide at the amino terminus.
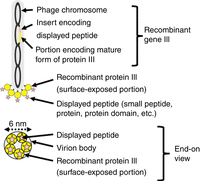
Schematic cartoons of virion structure, with a side view on top and an end-on view on the bottom. See text for further explanation.
I use the term “displayed peptide” (or simply “peptide”) generically for the amino acids encoded by the insert, regardless of the number of amino acids. So, for example, single-chain antibodies (∼240 amino acids; see Box 1) and short random peptides (typically 6–15 amino acids) are both called peptides, despite their disparity in size.
DISPLAY SYSTEMS
An important detail is suppressed in this and all upcoming cartoons for the sake of simplicity. In most affinity-selection projects, the recombinant gene III resides in a complete phage genome that bears no other gene III: a so-called type 3 display system (Smith 1993; Smith and Petrenko 1997). This system is well-adapted for short displayed peptides, which are likely to be displayed on all five recombinant pIII molecules at the tip of the virion. This is what is shown in Figure 1, and what is assumed here unless otherwise indicated. In many important projects, however, the displayed peptide is a ∼240-amino-acid single-chain antibody (Kang and Seong 2020) or other antibody domain; phages displaying such constructs are referred to generically as phage–antibodies. Almost all phage–antibody projects, along with other projects involving relatively large displayed peptides, use a type 3 + 3 display system (Smith 1993; Smith and Petrenko 1997) in which there are two gene III sequences in two different genomes: a special type of plasmid called a phagemid that bears the recombinant gene III encoding the displayed peptide and a helper phage that bears a wild-type or engineered gene III that does not encode a displayed peptide. A cell containing both genomes releases two types of virions: virions containing the phagemid chromosome and virions containing the helper genome. Both types of pIII molecules can be incorporated into both types of virions. When the displayed peptide is a relatively large domain such as a single-chain antibody, it is only occasionally expressed intact on either type of virion, for two reasons. First, the pIII encoded by the helper phage almost always outcompetes the recombinant pIII encoded by the phagemid for incorporation during virion assembly. Second, only a few large displayed foreign peptide domains escape degradation in the cytosol or periplasm during virion assembly, leaving only the rest of the protein intact in the completed virion. Consequently, only a minority of completed virions display an intact, functional foreign peptide domain; virtually no virions display two or more intact foreign peptide domains; and many virions display no intact foreign peptide domains at all. On the few occasions when antibody-bearing phagemid virions are mentioned in the following text, I assume (contrary to the cartoons) that an intact, functional antibody domain is displayed monovalently or not at all.
pIII molecules form a five-member ring that caps off the end of the long filamentous virion as it emerges from the cell (Rakonjac et al. 2017). This five-member ring is represented schematically in the end-view cartoon in the lower part of Figure 1; in the upper, side-view cartoon, it is represented less realistically as a fan. The carboxy-terminal half of the recombinant pIII is not depicted; it is buried within the tight architecture of the virion body and is not exposed to the surrounding medium. The amino-terminal half, with the displayed peptide at the very amino terminus, is flexibly attached to the virion, where it is exposed to the medium. The part of the exposed amino-terminal half that corresponds to wild-type pIII is represented by the yellow balls in both the side-view and end-view cartoons. The displayed peptide, represented here by the pink star, is flexibly attached to the phage-derived domain (the yellow ball), and is fully exposed to the medium. Virions belonging to other phage clones bear different gene-III inserts (they are colored differently in the following cartoons), and thus display different peptides (they have different shapes and the same color as the insert in the following cartoons).
There are two starting resources in an affinity-selection project:
-
A library with millions or billions of different phage clones, each displaying a different peptide (represented by different shapes with different colors in Fig. 2), and each represented by a relatively small number of identical virions. A large-scale affinity-selection project might start with 1 mL of buffer containing 1010 clones displaying 1010 different peptides, each represented by 1000 identical virions on average.
-
A selector, such as an antibody, antigen, or receptor molecule. In the case of phage–antibody libraries (see Box 1), for instance, the selector is a single antigen of interest. (In some projects, the selector is not a molecule. In other projects, the selector is a collection of thousands of different molecules represented in varying proportions—for example, all the molecules on the surface of some cell type. I will not consider such cases explicitly here.)
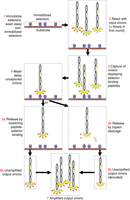
Schematic representation of the steps in a single round of affinity selection. The numbered steps are explained in the text.
The goal of affinity selection is to isolate rare library clones with virions displaying selector-binding peptides—for example, rare phage–antibody clones with virions displaying antibodies that bind the antigen acting as the selector.
The cartoons in Figure 2 show the steps in a single round of affinity selection; almost all affinity-selection projects include two or more successive rounds. The steps below are numbered to correspond to the step numbers in the figure. There are countless innovative variations on the basic procedure, some of which bear little resemblance to the cartoons. But the standard process covers the majority of affinity-selection experiments, and most of the principles here apply even to such procedural variants.
-
Step 1. The selector is immobilized on a solid surface that is called the substrate. Typical examples are the surface of a polystyrene Petri dish, the surface of the well in a multiwell polystyrene dish, and the impermeable hydrophilic surfaces of 1-µm paramagnetic beads (e.g., SpeedBeads from Cytiva Life Sciences).
Most protein selectors can be immobilized by direct absorption onto the polystyrene surface of a Petri or multiwell dish; this is thought to entail local denaturation of a small patch of protein surface, exposing hydrophobic groups that bond with the hydrophobic polystyrene. Alternatively, the selector can be covalently coupled to a hydrophilic surface derivatized with a reactive group such as amine-reactive N-hydroxysuccinimide or sulfhydryl-reactive N-alkyl maleimide; this method does not ordinarily entail denaturation of the selector.
A third, very popular immobilization mode is to react a biotinylated selector with a hydrophilic substrate coated with streptavidin or neutravidin (an engineered derivative of hen's egg avidin), each molecule of which can bind four biotin molecules; the selector is not ordinarily denatured. Small (1-µm in diameter) paramagnetic beads densely coated (∼0.2 molecules per nm2) with streptavidin or neutravidin are commercially available (SpeedBeads from Cytiva Life Sciences), as are 96-well dishes coated with streptavidin at ∼0.2 molecules per nm2 or with neutravidin at ∼0.024 molecules per nm2 (Thermo Fisher Scientific). Binding of biotin to streptavidin or neutravidin is extraordinarily fast, and dissociation is so slow that the bond is effectively covalent. The dissociation equilibrium constant, which is the ratio of the dissociation rate constant to the association rate constant, is roughly 1–10 fm. Essentially all selector molecules that are exposed to the surface are immobilized until the binding capacity of streptavidin or neutravidin is saturated. Streptavidin and neutravidin can themselves act as selectors, necessitating measures to avoid selecting unwanted virions that display streptavidin- or neutravidin-binding peptides. One such measure is to alternate streptavidin and neutravidin in successive rounds of affinity selection, because many peptides that bind one do not bind the other.
Whichever immobilization mode is chosen, the substrate must be thoroughly washed to remove all nonimmobilized selectors before proceeding.
-
Steps 2 and 3. The selector-coated substrate is reacted with the input virions. In the first round, the input virions are the library; in subsequent rounds, the input virions are the amplified output virions from the previous round (Step 7). During the reaction, virions displaying selector-binding peptides are captured on the substrate surface by immobilized selectors, whereas other virions (the overwhelming majority) remain free in solution. Often unwanted virions are also captured for reasons other than binding of their displayed peptides to the selector—for example, virions displaying peptides that bind streptavidin or neutravidin. These unwanted virions are examples of selector-unrelated phages (SUPs), which are the subject of a later section.
-
Step 4. The substrate surface is then thoroughly washed, with the aim of removing all virions that have not been captured—again, the overwhelming majority. The captured virions that remain after washing are ready for the next step: release from the substrate surface. I will discuss two alternatives for release.
-
Steps 5a and 6a. One common way of releasing captured virions is to elute them with an eluent that loosens peptide–selector binding without permanently damaging the virions. Examples of such eluents are acidic (e.g., 1 m glycine, pH adjusted to 2.2 with HCl) and basic (e.g., 100 mm triethylamine, pH 12) solutions. Once the eluate is transferred from the substrate to another vessel (Step 6a), the solution is readjusted to physiological conditions (e.g., neutral rather than acidic or basic pH) to allow the virions to recover infectivity and the displayed peptides to refold into their native conformation (if any). These are the unamplified output virions.
A potential disadvantage of release by loosening selector–peptide binding is that it is possible that virions displaying peptides with particularly high affinity for the selector might be particularly resistant to release. This is obviously very undesirable if the goal is to discover peptides with particularly high affinity for the selector. It should be noted, however, that there is little danger of this undesirable bias when acid or alkali is used to loosen the binding between a virion-borne, single-chain antibody and an immobilized antigen selector, or between a virion-borne peptide and an immobilized antibody selector. This is because acid or alkali loosens antigen–antibody binding not only directly, by interfering with the molecular interactions between the antibody's binding site and the antigen's epitope (the part of the antigen that makes direct contact with the antibody's binding site), but also indirectly, by loosening the three-dimensional folded framework that holds the segments of the antibody's antigen-binding site together.
-
Steps 5b and 6b. A second common way of releasing captured virions is cleaving the peptide from the virion with the intestinal protease trypsin (Thomas and Smith 2010). This requires that one or more trypsin-sensitive bonds (lysine or arginine followed by any amino acid other than proline) be engineered or exist naturally between the peptide and pIII. Most single-chain antibody domains (see Box 1) have a trypsin-sensitive bond at the carboxy-terminal end. Filamentous virions, the natural habitat of which is the mammalian intestine, are themselves highly resistant to trypsin and other intestinal proteases (Salivar et al. 1964; Sieber et al. 1998). (The cartoon for Step 5b shows the detached peptides intact in solution or still bound to the selector, but either peptide or selector or both may also be cleaved by trypsin.) Once the released virions are transferred to another vessel (Step 6b), trypsin can be neutralized with a commercially available trypsin inhibitor (e.g., lima bean trypsin inhibitor from Worthington Biochemical). These are the unamplified output virions; unlike the unamplified output virions in Step 6a, they are “denuded” (stripped of their displayed peptides).
A key advantage of trypsin release is that it does not depend at all on the nature and strength of peptide–selector interaction. Therefore, there is no possibility of bias that depends on the strength or nature of that interaction—a distinct possibility when release is accomplished by loosening peptide–selector interaction, as explained in the text for Steps 5a and 6a. Another advantage is that denuded virions have uniform, relatively high infectivity (ability to infect susceptible cells); this means that they are approximately uniformly represented in the first cohort of infected cells when the unamplified output virions are amplified (Step 7). In contrast, large displayed peptides can have large and highly variable inhibitory effects on infectivity (Løset et al. 2008).
-
Step 7. The unamplified output virions (Step 6a or 6b) are used to infect fresh bacterial host cells, which are then cultured, producing enormous numbers of copies of each of the infecting virions. This process is called amplification, and the resulting population constitutes the amplified output virions. Amplification is absolutely necessary to prepare output virions in sufficient numbers for another round of affinity selection or initial analysis of the output clones—what I will call readout. For the purposes of readout, amplification is often performed with individual output clones rather than en masse. In a later section (“NGS as Primary Readout”), however, I will advocate for en masse readout.
YIELD VERSUS STRINGENCY
Yield is the probability that a virion from a given clone in the input will be represented in the unamplified output. Technically, it is an abstract property intrinsic to the clone under specified conditions of affinity selection; the actual ratio of unamplified output virions to input virions is randomly distributed about the intrinsic yield.
It is important to distinguish between specific yield, which is highly dependent on the identity of the displayed peptide, and nonspecific background yield, which is largely independent of the identity of the displayed peptide. The obvious reason for a relatively high specific yield is that the clone in question displays a peptide with relatively high affinity for the selector. Such specific clones are called selector-specific, as are their yields. But some specific clones owe their relatively high specific yields to something other than the affinity of their displayed peptides for the selector. Such clones are examples of SUPs, which are the subject of the next section.
Nonspecific background yield is never zero (no selection process is perfect), but is generally very low. It can depend on the physical setup (e.g., the geometry and composition of the substrate) and the details of the selection procedure. Unless selector-specific yield is considerably higher than nonspecific background yield, there is no way to distinguish the desired selector-specific clones, which are very rare in the starting library, from the background clones, which are overwhelmingly abundant in the starting library, and which emerge randomly from imperfection of the selection process.
Stringency is the degree to which affinity selection favors virions with high selector-specific yield over those with low selector-specific yield (I will not attempt an algebraic definition). High stringency too is desirable.
Increasing stringency generally reduces selector-specific yield relative to nonspecific background yield. Conversely, increasing selector-specific yield generally reduces stringency and may also increase nonspecific background yield. Choosing which desirable characteristic to emphasize is a critical strategic consideration in the design of the affinity-selection projects.
-
In the first round of affinity selection, high selector-specific yield is of the highest priority, even though that means sacrificing stringency. The reason is that the input library contains a very large number of clones, each represented by relatively few virions. Consider one of the desired clones: a clone with a displayed peptide that binds the selector with high affinity. If we fail to capture at least one of that clone's virions in the first round, the clone obviously cannot reappear magically in subsequent rounds.
-
In the second and subsequent rounds of affinity selection, selector-specific yield can be sacrificed in the interest of high stringency. This is because there are orders of magnitude fewer input clones in these rounds, and each clone is represented by orders of magnitude more virions. If selection is too stringent, however, clones that appear in the output because of their peptides’ high affinity for the selector cannot be distinguished from nonspecific background clones that appear randomly in the output because the selection process is not perfectly discriminatory.
The obvious way to maximize selector-specific yield in the first round is to maximize the surface density of the selector on the substrate. This promotes high yield in two ways. First, the higher the surface density of selector, the faster the rate at which virions displaying selector-binding peptide will be captured on the surface, and the more likely that virions that disengage from the surface will reengage.
But there is a second important way that high selector surface density increases selector-specific yield: the avidity effect. Recall that five copies of the displayed peptide form a ring at one tip of the virion (cartoon in the lower part of Fig. 1). If the selector is densely arrayed on the substrate surface, it is possible for two peptides on a single virion to bind simultaneously to two neighboring immobilized selectors: bivalent binding. Bivalent binding greatly increases the strength with which the virion is captured on the substrate surface. This is because, if one peptide temporarily disengages from its selector, the virion is not free to diffuse away because it is held in place by the other peptide–selector interaction; the temporarily disengaged peptide is thus in position to rapidly reengage its selector. The result is that the virion's effective dissociation rate from the substrate is orders of magnitude slower than the underlying monovalent dissociation rate of an individual peptide from an individual selector molecule.
The avidity effect is largely irrelevant in the case of single-chain antibodies (and most other protein-sized peptides) displayed on phagemid virions. This is because display of these large “peptides” is almost always monovalent (Box 1).
How can stringency be increased in the second and subsequent rounds? Three approaches are considered.
-
The obvious approach is to reduce the surface density of the selector. This will favor displayed peptides with fast association rates and slow dissociation rates. It will also tend to decrease the avidity effect. The avidity effect is a disadvantage when stringency is at a premium, because even virions displaying peptides with weak monovalent affinity for the selector can be strongly captured on the substrate when two of their peptides are bound to selectors. Avoiding the avidity effect thus favors displayed peptides with slow monovalent dissociation rates. But there is a limit to how far stringency can be increased before selector-specific yield descends to the level of nonspecific background yield. For some libraries and selectors, it may not be possible to reduce surface density sufficiently to avoid the avidity effect without erasing the difference between selector-specific and nonspecific background yields. This is an issue that is addressed in the section on “How to Regulate Stringency.”
-
A second approach is to use stringent washing conditions (Step 4 in Fig. 2) that partially loosen the binding between displayed peptides and the selector. The hope is that virions displaying weakly binding peptides will be preferentially washed away, whereas virions displaying strongly binding peptides will remain captured on the substrate surface. Perhaps this can be accomplished simply by longer and more vigorous washing. Alternatively, the substrate can be washed with nonphysiological solutions that partly loosen peptide–selector binding. It should be borne in mind, though, that the peptides with binding favored under such nonphysiological conditions may not be the same as the peptides with the strongest affinity under physiological conditions.
-
A third approach is competition with a known selector ligand. This option is available only in very special circumstances and will not be discussed further.
THE SCOURGE OF SUPs
A selector-unrelated phage (SUP) is a phage clone that increases in prevalence over the course of affinity selection for some reason other than binding of its displayed peptide to the selector (Menendez and Scott 2005; Brammer et al. 2008; Thomas et al. 2010). Many affinity-selection projects are plagued by such SUPs.
SUPs have previously been called TUPs, which originally stood for target-unrelated peptides. There are two problems with this nomenclature. First, “target” is an ambiguous term for the selector, because it might equally be taken to refer to the peptide. Second, the problem may not have anything to do with a clone's displayed peptide, as will be explained below.
There are two categories of SUPs (some SUPs may fall into both categories):
-
Capture- or release-related SUPs. Their increased prevalence reflects increased yield in the unamplified output virion population (Step 6a or 6b in Fig. 2) relative to the input population (Step 2). Such increased prevalence presumably involves some characteristic of the displayed peptide, as this is the only difference between clones at this stage; it is thus an example of high specific yield, but it does not depend on the identity of the selector. Perhaps the peptide binds the substrate or some other element of the selection apparatus. A common example of capture-related SUPs are clones displaying peptides that bind streptavidin or neutravidin when these biotin-binding proteins are used to mediate immobilization of biotinylated selectors (see the explanation for Step 1 in Fig. 2).
-
Propagation-related SUPs. Their increased prevalence reflects overrepresentation in the amplified output virion population (Step 7 in Fig. 2) relative to the unamplified output virion population (Step 6a or 6b). Overrepresentation could reflect increased infectivity, increased replication inside the infected cell, increased growth of infected cells, or increased production of completed virions in the medium. Some propagation-related SUPs can be related somehow to the displayed peptide. As a particularly perverse example, in type 3 + 3 systems (see Box 1), deletion or lack of expression of the recombinant gene III (which encodes the peptide) can give the infected cell a growth advantage, if that particular recombinant protein is somewhat toxic. Other propagation-related SUPs are unrelated to the displayed peptide. For example, the clone may harbor a mutation elsewhere in the genome that increases replication of phage DNA in the infected cell (Brammer et al. 2008; Thomas et al. 2010).
There are several countermeasures that can be taken against SUPs:
-
Unnecessary propagation steps should be avoided to minimize propagation-related SUPs (Thomas et al. 2010). Of course, some propagation steps are unavoidable, including amplification of unamplified output virions. But others are avoidable, including, for example, multiple serial reamplifications of the library. Obviously, avoiding unnecessary propagation can have no effect on capture- or release-related SUPs.
-
Counterselection with a mock selector (or no selector at all) may remove unwanted virions that bind to some element of the selection apparatus other than the selector (for example, the substrate). The virions that escape capture in the counterselection (Step 3 of Fig. 2) are retained; there is no need for the subsequent steps depicted in Figure 2. Meanwhile, the counterselected virions serve as input to the subsequent round of ordinary (positive) affinity selection (Steps 2 and 3). Obviously, counterselection can only affect capture- or release-related SUPs; it can have no effect on propagation-related SUPs.
A common example is counterselection with streptavidin and/or neutravidin in the absence of biotinylated selector when those biotin-binding proteins are used to mediate immobilization of biotinylated selectors for the subsequent round of positive selection (see the explanation for Step 1 in Fig. 2). This counterselection is a useful complement to alternating between streptavidin and neutravidin in successive rounds of positive selection.
Counterselection is almost never 100% effective; two or more consecutive rounds of counterselection are typically required to sufficiently reduce the concentration of unwanted SUPs. It should also be borne in mind that, in many cases, especially with large displayed peptides, counterselection cannot permanently remove undesired clones from the phage population. This is because some virions representing such an undesired clone may not display the unwanted peptide in intact form, and thus will not be subject to counterselection. In such cases, amplification subsequent to counterselection will partially restore representation of the unwanted clone.
-
SUPs may be identified bioinformatically without being removed. Thus, a series of control affinity selections with a control selector (or no selector at all) is performed in parallel with the real series of positive affinity selections, using exactly the same procedures in both cases. The frequencies of the clones that emerge from each selection are compared. Clones that are abundant in the output of both selection series are SUPs; both capture- or release-related and propagation-related SUPs can be detected in this way. Clones that are abundant in the output of the positive selection series but rare in the output of the control selection series may well display a selector-binding peptide—exactly what is sought in an affinity-selection project. Clones that are rare in the output of the positive selection series but abundant in the output of the control selection series may display a peptide that binds the control selector (if any). This approach requires analyzing large numbers of output clones, which is one of the arguments for next-generation sequencing (NGS) as the primary readout in affinity-selection projects, the subject of the next section.
NGS AS PRIMARY READOUT
Next-generation sequencing (NGS) has become an essential tool in life sciences today. In the context of affinity-selection projects, it can provide a thorough census of the sequences of the displayed peptides, including single-chain antibodies, in the output virion population of an affinity-selection project (Brinton et al. 2016; Yang et al. 2017; Rouet et al. 2018; Braun et al. 2020; Zambrano et al. 2022). Tens or even hundreds of millions of output virion sequences from positive and control affinity selections become available for bioinformatic analysis. These results allow enumeration of the number of times that the sequence from any given clone appears among those tens or hundreds of million sequences; this number is called the clone's reads. The number of a clone's reads divided by the total number of reads is a measure of the clone's prevalence in the population. Such population prevalence data can pinpoint clones that are particularly likely to display peptides with high affinity for the selector, as explained in this section. Moreover, the displayed peptide sequences can be subjected in turn to further bioinformatic analysis—for example, to identify common sequence motifs (O'Shea et al. 2013). For these reasons, NGS is increasingly favored as the primary readout in affinity-selection projects: the first level of analysis of output clones.
NGS delivers sequence information, not physical clones. But modern molecular biology provides abundant means for using sequence information to recover any particular clone of interest from the output phage population for further analysis and development.
NGS cannot deliver a census of a large starting library with billions of clones, each displaying a different peptide. But affinity selection reduces the number of clones so drastically that 10 million NGS reads will ordinarily provide a nearly complete census of the first-round amplified output, and thus a measure of each clone's prevalence in that output population.
First-round prevalence data by themselves do not suffice to identify promising clones for two reasons. First, only for a tiny minority of the clones in the first-round amplified output can their prevalence in that output be compared to their prevalence in the first-round input (the large starting library); thus, the yield during the first round of affinity selection (essential for assessing the promise of a clone's displayed peptide as a high-affinity ligand for the selector) cannot be calculated except in extremely rare cases. Second, as explained in the section on “Yield Versus Stringency” above, the first round of affinity selection must prioritize yield, severely compromising its ability to discriminate among clones on the basis of the affinity of their displayed peptides for the selector.
The full value of a thorough first-round census becomes apparent only when a thorough second-round census is also available. This allows each clone's yield in the second round of affinity selection to be estimated by comparing its prevalence in the second-round output population to its prevalence in the second-round input population (the same as the first-round amplified output population). If, again following the principles outlined in the section on “Yield Versus Stringency” above, the stringency of the second round has been adjusted such that the yields of selector-specific clones reflect the affinity of their displayed peptides for the selector, this information suffices to identify clones that are promising candidates for further investigation.
It may be necessary in a few cases to analyze the third or even fourth round of affinity selection. Eventually, however, information will decline in succeeding rounds, as clones with the highest yield crowd out clones with lesser yields. This is a severe disadvantage even if the goal of the project is to identify the clone with the highest affinity for the selector. This is because the specific clones with the very highest yields may be undesired SUPs rather than the desired clones displaying peptides with high affinity for the selector. It is vital to collect data for all clones with specific yields substantially higher than the nonspecific background yield.
Figure 3 is a schematic diagram showing NGS analysis in the context of a two-round affinity-selection project from a type 3 + 3 phage–antibody library (see Box 1): for instance, a library displaying single-chain antibodies (Andris-Widhopf et al. 2000) derived from chicken immunoglobulin Y (Carlander et al. 2000). The recombinant pIII displaying the single-chain antibody domain is encoded on a phagemid chromosome, which also bears a phage origin of replication and a selectable marker (typically ampicillin resistance), but no additional phage elements. Only when phagemid-bearing cells are superinfected with helper phage virions, which encode an alternative pIII (either wild-type or engineered) as well as all the other phage proteins necessary for virion assembly, do the cells produce virions: both phagemid and helper virions. In phage–antibody projects, it is the antigen of interest that serves as the selector. The steps highlighted in red in Figure 3 are discussed below.
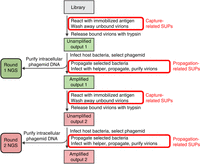
Flow diagram for two rounds of affinity selection, starting with a library of single-chain antibodies displayed from a phagemid vector. The output of each round is analyzed by next-generation sequencing (NGS). Further explanation can be found in the text. (SUPs) Selector-unrelated phages.
In each round, the input (the initial library in round 1 and the amplified first-round output in round 2) is a mixture of phagemid and helper phage virions (the former predominating because of the design of the helper). Some of each type of virion display a single functional single-chain antibody; almost no virions display more than one. Capture and release (Steps 1–6a or Steps 1–6b in Fig. 2) proceed as usual, resulting in the unamplified output (Step 6a or 6b).
Amplification in type 3 + 3 systems is broken down into distinct steps. First, the output is used to infect cells. Second, the infected cells are cultured under conditions that select for the phagemid's selectable marker (again, usually ampicillin resistance). When the culture has grown sufficiently to ensure that all infecting phagemids are abundantly represented in the population, part of the culture is split off and allowed to continue growth; this culture is the source of the phagemid DNA that is subjected to NGS, as symbolized in the figure by the leftward-branching arrows. Meanwhile (third), the remainder of the phagemid culture is superinfected with a large excess of fresh helper virions, and the doubly infected cells are cultured to yield the amplified output virions: both phagemid and helper phage, with the former predominating. As usual, the amplified output virions from the first round are the input virions for the second round.
The red rectangles in Figure 3 highlight the steps in each round at which bias in favor of SUPs can occur:
-
The capture stage of affinity selection can favor capture-related SUPs—for example, virions with a displayed peptide that binds some component of the affinity-selection apparatus other than the selector.
-
If release is accomplished by trypsin cleavage, there is little room for bias of any kind, including bias in favor of SUPs during the release stage.
-
As trypsin release results in denuded virions (including phagemid virions) in the unamplified output, there is also little room for bias during infection of bacterial cells with phagemid virions.
-
However, there is opportunity for bias in favor of propagation-related SUPs during propagation of phagemid-infected cells and during virion assembly after superinfection with helper virions.
Because bias in favor of SUPs is independent of the selector (the antigen of interest in the case of phage–antibody libraries), the very same SUPs should be favored in parallel affinity selections from the same initial library with different antigens as selectors. Comparisons of the clones emerging from the parallel selections should allow likely SUPs of all kinds to be identified, as explained in the section on “The Scourge of SUPs” above. In light of this consideration, it makes sense to carry out affinity selections in batches, with each affinity selection in a batch using a different selector. Apart from the choice of selector, the different affinity selections should be as identical as possible. That way, the different affinity selections serve as controls for one another.
HOW TO REGULATE STRINGENCY
The section on “Yield Versus Stringency” above explains that stringency can be increased in the second and subsequent rounds of affinity selection by decreasing the surface density of the selector. This favors virions that are captured at a fast rate, and reduces the avidity effect, which would allow unwanted virions displaying peptides with weak monovalent affinity for the selector to contribute to the output. However, there is a limit to this approach. It may not be possible to reduce selector surface density sufficiently to significantly discourage the avidity effect without simultaneously severely reducing the ratio of specific yield to nonspecific background yield. If the ratio is reduced too much, the desired clones (those displaying selector-binding peptides) are barely distinguishable in frequency from random background clones that appear only because the selection process is not perfectly discriminatory.
This is not a problem for phage–antibody libraries (Box 1), in which display is monovalent and avidity is therefore irrelevant. This section applies instead to projects in which five peptides are displayed on each virion, as is likely to be the case for type 3 libraries (Box 1) displaying short peptides.
This section outlines a possible alternative selection process in which the avidity effect might actually be exploited to enhance selection in favor of clones with displayed peptides that bind the selector with particularly strong monovalent affinity. It should be kept in mind that the proposal is speculative at this time.
The proposed alternative affinity-selection process differs from the usual one in the capture stage (Steps 1–3 in Fig. 2). It begins by reacting the library with selector molecules free in solution, not immobilized on a substrate. During this liquid-phase reaction, selectors bind reversibly to displayed peptides; the system is allowed to come to equilibrium.
The selector molecules are assumed to have been biotinylated. As described in the explanation for Step 1 in Figure 2, biotinylated molecules bind streptavidin or neutravidin with an extraordinarily fast association rate and an extraordinarily slow dissociation rate—exactly what is required for the next stage of the proposed procedure.
The next stage is to remove unbound selectors (biotinylated selector molecules that are not bound to displayed peptides) and immediately deliver the virions, along with their still-bound selectors, directly onto a substrate surface that is densely coated with streptavidin or neutravidin, where they are rapidly captured on the substrate surface. This might be accomplished in a matter of minutes (before selectors that are strongly bound to displayed peptides have a chance to dissociate) by gel filtration through a spin column from which the effluent flows directly into a suspension of streptavidin- or neutravidin-coated 1-µm paramagnetic beads (e.g., SpeedBeads from Cytiva Life Sciences; 0.2 streptavidin or neutravidin molecules per nm2).
Spin columns (for example, Zeba desalting columns from Thermo Fisher Scientific) are widely used in molecular biology to remove small solutes from macromolecules such as proteins and nucleic acids. In that case, the gel filtration medium is beads with small pores that only small solutes can penetrate, allowing macromolecules to emerge from the column free of these solutes. Because the solution is driven through the column by centrifugal force, filtration is finished in a few minutes. In the process proposed here, the gel filtration medium would be beads such as Sephacryl S-500 HR (MilliporeSigma) with pores that allow macromolecules such as selectors to penetrate, whereas particles like virions are excluded (Zakharova et al. 2005). It remains to be determined whether such a column would suffice to free virions from almost all selectors that are not bound to displayed peptides.
From this point on, the proposed alternative affinity-selection procedure is the same as the usual procedure (Steps 4–7 in Fig. 2). In particular, the substrate is thoroughly washed to remove virions that are not captured on the surface. The substrate is then ready for the usual release stage, either by loosening peptide–selector binding (Steps 5a and 6a in Fig. 2) or by trypsin cleavage (Steps 5b and 6b), as described in the section “The Affinity-Selection Process.”
The attraction of the proposed alternative capture procedure lies in the kinetics of reversible solution-phase selector–peptide binding. It is to those kinetics that I now turn, making the following simplifying assumptions:
-
Selectors bind monovalently to displayed peptides. Either one or two selectors can be bound to a single virion; in the latter case, the two selectors are bound to nonvicinal peptides (see bottom cartoon in Fig. 1).
-
All selector–peptide interactions for a given clone occur independently with the same reversible monovalent kinetics.
-
All reversible-binding interactions for a given clone are governed by the same monovalent kinetic parameters:
-
– an association rate constant (dimensional units time−1 concentration−1),
-
– a dissociation rate constant (dimensional unit time−1), and
-
– the dissociation equilibrium constant KD, which is the dissociation rate constant divided by the association rate constant. KD has the dimension unit of concentration.
-
At selector concentrations at which the selector can plausibly bind to a displayed peptide at significant levels, the selector will be in vast molar excess over all selector-binding peptides. Effectively, therefore, the free selector concentration can be assumed to be the same as the total selector concentration, which is abbreviated as “S.”
Each virion is assumed to display five peptides in a ring at one tip of the particle (lower part of Fig. 1). There are therefore five ways that a virion can associate with a selector, and only one way for a virion with one bound selector to dissociate. The overall dissociation equilibrium constant for virions with no versus one bound selector will thus be the monovalent dissociation rate constant divided by five times the monovalent association rate constant, or KD/5. Similarly, there are two ways that a virion with one bound selector can associate with another selector to create a virion with two bound selectors, and there are two ways that a virion with two bound selectors can dissociate to create a virion with one bound selector. The overall dissociation equilibrium constant for virions with one versus two bound selectors will thus be two times the monovalent dissociation rate constant divided by two times the monovalent association rate constant, or KD. The two types of reversible binding reactions are diagrammed in the upper part of Figure 4.
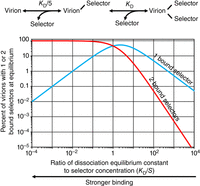
(Top) Schematic representation of the equilibria governing the reaction of the selector with a single-phage clone with a displayed peptide that binds the selector with a monovalent dissociation equilibrium constant of KD. The simplifying kinetic assumptions are explained in the text. (Bottom) Graph showing the fraction of a clone's virions with one or two selector molecules bound to the virion's displayed peptides at equilibrium, as a function of the ratio of the dissociation equilibrium constant for these peptides to the concentration of the selector. The simplifying kinetic assumptions underlying the calculation are explained in the text.
At equilibrium, the equilibrium equations for both reversible reactions (between virions with no bound selector and virions with one bound selector [governed by the dissociation equilibrium constant KD/5] and between virions with one bound selector and virions with two bound selectors [governed by the dissociation equilibrium constant KD]) must be simultaneously satisfied for every phage clone displaying a selector-binding peptide.
Let us consider a single phage clone with a displayed peptide that binds the selector with a monovalent dissociation equilibrium constant of KD (the lower the value of KD, the stronger the binding affinity), and define the ratio R ≡ KD/S, the peptide's dissociation equilibrium constant relative to the prevailing selector concentration. In this case, following the simplifying kinetic assumptions and their consequences in the preceding paragraphs, we can calculate the fraction of that clone's virions that have a selector molecule bound to one of its displayed peptides at equilibrium as 5R/(5 + 5R + R2). Similarly, the fraction of the clone's virions with selector molecules bound to two of its displayed peptides at equilibrium is 5/(5 + 5R + R2). The graph in the lower part of Figure 4 plots the percent of a clone's virions with one or two bound selectors as a function of R, equal to that clone's peptide dissociation equilibrium constant KD divided by the selector concentration S.
Once equilibrium has been established, the solution is processed using the alternative capture phase presented earlier in this section. During this process, almost all free biotinylated selector molecules will (hopefully) be removed by the spin column before the solution arrives at the streptavidin- or neutravidin-coated substrate. Virions with peptides that have high dissociation rate constants will lose one or both of their bound selectors to the gel filtration medium in the spin column. But the most desirable virions—those with one or especially two selectors still bound to their peptides—will be captured monovalently or bivalently on the substrate surface. If that substrate surface is then washed vigorously, it may be possible to eliminate most virions that are not captured bivalently through two bound selectors. As the red curve in the graph indicates, this will result in sharply progressive discrimination against virions with displayed peptides that have monovalent dissociation equilibrium constants KD progressively greater than the prevailing selector concentration S. In this context, the avidity effect from bivalent binding works for, rather than against, the goal of discovering peptides with strong monovalent binding to the selector.
The effectiveness of this approach depends critically on nearly complete removal of free selector molecules by gel filtration through spin columns. Whether this is possible with macromolecular solutes, with their small diffusion constants, has yet to be addressed to my knowledge.
Footnotes
-
From the Advances in Phage Display collection, edited by Gregg J. Silverman, Christoph Rader, and Sachdev S. Sidhu.








