
Studying Mitosis in Cultured Mammalian Cells
This protocol was adapted from “Studying Mitosis in Cultured Mammalian Cells,” Chapter 27, in Live Cell Imaging (eds. Goldman and Spector). Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, USA, 2005.INTRODUCTION
A major challenge for those who study mitosis is the need to keep cells alive and mitotically active during observations. This protocol describes methods for maintaining healthy, dividing mammalian cells in culture and during imaging, when mitosis can be examined. Rose chambers are preferable for observation and microinjection of living mitotic cells, but slide/coverslip preparations are easy to make and do not require any special equipment. Another inexpensive and easy-to-use alternative is to grow cells in a culture dish with a glass bottom. Such dishes are suitable for microinjection experiments.
RELATED INFORMATION
The process of mitosis is perhaps best appreciated from movie sequences illustrating the dynamic nature of the mitotic process.Movie 1 shows mitosis in pig kidney cells expressing GFP-tubulin. The sequence shows a cell from prophase through cytokinesis.
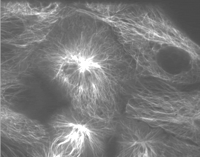
Mitosis in pig kidney cells expressing GFP-tubulin. The sequence shows a cell from prophase through cytokinesis.
MATERIALS
Reagents
Cells to be examined
Detergent (Alconox)
Fetal calf serum (optional; see Step 1)
Medium, appropriate for the cells being cultured (as recommended by the American Type Culture Collection, bicarbonate-buffered, supplemented with 10% fetal calf serum and antibiotics)Medium, same as the above, except buffered with HEPES, 20 mM, pH 7.2 instead of bicarbonate)OptiMEM (Invitrogen) (optional; see Step 1)
Oxyrase (optional; see Step 2)
Equipment
Bunsen burner
Coverslips, glass
Coverslips, gridded (Bellco Glass) (optional; see Step 4)
Culture dish with a glass (coverslip) bottom (for glass-bottomed culture dish method)
Hot plate (for slide/coverslips method)
Incubator preset to 37ºC; 5%-10% CO2
Microscope (inverted or upright, depending on method used for mounting coverslips)
Paintbrush, small (for slide/coverslips method)
Parafilm (for slide/coverslips method)
Rose chamber (optional; see Step 15)
Slides, glass
Tissues
METHOD
Growing Cultured Cells
General procedures for tissue culture, including basic equipment needed in a culture laboratory, sterile technique, media preparation, and procedures for subculturing, freezing, and thawing cells can be found in many laboratory methods books (e.g., Barker 1998).
-
Table 1 compares some key features of mitosis in various cultured cell lines.
-
1. Plate cells 1-3 days before use in medium that is buffered with bicarbonate and supplemented with 10% fetal calf serum and antibiotics. Grow cells in 5%-10% CO2 at 37ºC in a humid atmosphere.
In some cases, medium that has been mixed 1:1 with OptiMEM prepared with 5% serum improves cellular morphology.
Maximize the number of cells in mitosis at the time of observation by plating the cells densely enough so they are in exponential growth at the time of the experiment. Determine the plating density empirically for each cell line.
-
2. For observation of living cells in mitosis, remove the bicarbonate-buffered medium and replace it with HEPES-buffered medium.
Although cells proliferate faster in CO2-containing medium, maintaining the cells in HEPES-buffered medium for short-term observations (several hours) causes no detectable adverse effects. For fluorescence observations, use medium that lacks the pH indicator dye, which is autofluorescent. To reduce photobleaching during fluorescence observations, add the oxygen-scavenging reagent Oxyrase at a dilution of 1:100 in the medium.
-
3. For long-term storage, freeze cultured cells in the bicarbonate-buffered medium used for growing cells in Step 1. Supplement the medium with 15% DMSO and 20% serum. Store the frozen cells in liquid nitrogen tanks.
Mounting Coverslips for Imaging
-
4. Clean and sterilize coverslips as follows:
-
i. Hand wash coverslips in Alconox detergent.
-
ii. Rinse coverslips exhaustively in running hot H2O, and then rinse in distilled H2O.
-
iii. Store washed coverslips in 95% ethanol and flame sterilize before use.
-
-
An alternative, although less preferred, method for cleaning coverslips is to rinse them in 95% ethanol and flame sterilize before use. For microinjection experiments or for experiments in which cells are to be fixed and relocalized after imaging, grow cells on gridded coverslips.
Presented below are three ways to mount coverslips for imaging.
A. Slide/Coverslips
-
5. Plate cells on clean glass coverslips and allow them to grow to the desired density
-
6. Prepare parafilm spacers by cutting two thin strips of parafilm (~2 × 20 mm). Place them parallel to each other, about a coverslip’s width apart, on a clean glass slide.
Parafilm spacers are used to hold the coverslip above the slide surface and prevent the cells from getting crushed under the weight of the coverslip.
-
7. Remove a coverslip from the culture dish and blot excess medium onto a tissue.
-
8. Place a drop of medium between the parafilm spacers and carefully invert the coverslip onto the spacers.
-
9. Use the tip of a tissue to remove excess medium from the top of the coverslip and the surface of the slide.
-
10. Warm Valap at a low setting on a hot plate.
-
11. Use a small paintbrush to seal the coverslip to the slide with liquified Valap to prevent evaporation of medium during observation on the microscope.
B. Glass-Bottomed Culture Dishes
-
12. Seed the cells directly in a culture dish with a glass (coverslip) bottom.
-
13. Allow the culture to grow until the cells are of the desired density.
-
14. Change the medium to non-CO2 medium. Move the preparation to the stage of an inverted microscope.
These dishes can also be used with an upright microscope with a water-immersion objective lens. Glass-bottomed dishes are available with a gridded bottom (e.g., MatTek) for following individual cells over long periods. The plastic lid of the dish will interfere with polarized light microscopy, but it can be removed and replaced with a round, glass coverslip of suitable diameter.
C. Rose Chambers
-
15. A Rose chamber (Fig. 1) consists of a lower metal plate with a circular cutout, covered with a layer of parafilm to prevent the coverslip from cracking. The coverslip (with cells) is centered over the circular cutout and covered with a rubber gasket, a second coverslip, and the top metal plate. The entire assembly is held together by four screws. Medium can be added using a syringe and needle inserted into the gasket. Alternatively, the chamber can be constructed without the second coverslip; medium can then be added from above and the top opening of the chamber subsequently covered with a circular coverslip of 25-mm diameter.
 Figure 1.
Figure 1.Photograph and diagram of a Rose chamber used for imaging living cells. The photograph shows the assembled chamber; the coverslip with cells is on the bottom, and the chamber is used on an inverted microscope. The diagram shows the parts of the chamber. The upper and lower plates are aluminum, and the inner spacer is made by polymerizing a sheet of silastic elastomer of the desired thickness and cutting the openings using a cork borer. To prevent cracking the coverslip when the chamber is assembled, a piece of parafilm (with a central opening) can be placed over the lower plate before adding the coverslip. The entire assembly is held together with four screws, and a round coverslip is added as a lid on the top.
Although Rose chambers are not commercially available, several manufacturers make chambers similar in design, and some also can be used for perfusion experiments (Bioptechs). Rose chambers can be used on an inverted microscope or constructed with a thinner gasket and used on an upright microscope. Alternatively, the chamber can be inverted (or constructed with the cell-containing coverslip on the top) for use with an upright microscope. Rose chambers can be held on the stage of most microscopes, or specially designed stage inserts can be made.
DISCUSSION
The Rose chamber has many advantages. It is virtually leak-proof. It holds a larger volume of medium than a slide/coverslip preparation (the volume depends on the thickness of the rubber gasket), and this extra medium seems to keep the cells healthy for longer periods. The volume of the Rose chamber and the metal plates may help to reduce fluctuations in heating during observation. Because the chambers are sealed, they can be returned to the 37ºC incubator between observations or experiments. Coverslips mounted in a Rose chamber can be used for microinjection and subsequent high-magnification observations (the top coverslip is simply removed during injection and replaced when the cells are ready for imaging). Cells can be rapidly fixed after live cell observations by removing the top coverslip and adding fixative.
TROUBLESHOOTING
Problem: It is difficult to maintain the temperature of the cells.
[Step 2]
Solution: For observations of mitosis, the cells must be kept warm, ideally at 37ºC. For short-term studies, use an air-curtain-type incubator, which blows warm air at the sealed preparation. Insert a thermister probe directly into the Rose chamber to monitor the temperature of the preparation during heating. To avoid focus fluctuation, heat the microscope area before beginning the experiment. For long-term studies or for experiments that require more precise temperature control, use a chamber that surrounds the microscope or the stage. These are commercially available (e.g., Buck Scientific) or can be built from acrylic glass (e.g. Plexiglas) or plywood. Heated stages are available for some microscopes. If oil immersion is to be used, purchase (Bioptechs) or make a heating collar that surrounds the objective lens. (A lens heater can also be used in conjunction with the air-curtain-type incubator.) A heating block that holds a Rose chamber has also been designed. A disadvantage of all of these methods is the introduction of strain into the lenses by the repeated cycles of heating and cooling. Circumvent this problem by keeping the microscope lens in a dry incubator at 37ºC (Bioptechs) when not in use. A final alternative is to place the entire microscope in an environmental room of constant temperature.
Problem: It is difficult to maintain the cells in mitotic phase.
[Step 2]
Solution: In some cases, cells in early mitosis revert to interphase. During microinjection, this is thought to occur when calcium in the medium enters the cell. Removal of calcium from the culture medium can eliminate this problem, but the cells eventually detach from the surface. Prophase cells may also revert to interphase if they are damaged by light. Limit the exposure of the cells to light, especially light at shorter wavelengths. When viewing cells with tungsten light for phase contrast or differential interference contrast microscopy, use green light and/or add a heat cut filter. For fluorescence observations, expose cells to the least amount of fluorescent light possible.
Problem: Cell synchronization is needed.
[Step 2]
Solution: When a large number of mitotic cells needs to be followed or a particular mitotic stage is desired, synchronization can be useful. For example, prophase cells constitute a minority of the mitotic cells on a coverslip, but incubation of coverslips of cells for 90 minutes in nocodazole (10 μM) followed by 90 minutes of washout results in a transient increase in prophase cells in the population. Mitotic shake-off is an effective method to obtain relatively large numbers of cells in mitosis, but it is limited to cells (e.g., HeLa) that round up in mitosis. For cells that remain adherent during mitosis, use thymidine to block cells at the entry into S phase. This procedure is most effective when it is repeated (a double thymidine block). Use Colcemid after the removal of thymidine to obtain cells synchronously entering but blocked in mitosis. A disadvantage of these methods is that, depending on the length of the cell cycle, it can take several days to obtain a synchronized population of cells.








