
Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): Overview
Adapted from “DNA Linear Amplification,” Chapter 7, in Whole Genome Amplification: Methods Express (eds. Hughes and Laskin), from the Methods Express series. Scion Publishing Ltd., Oxfordshire, UK, 2005.INTRODUCTION
T7-based linear amplification of DNA (TLAD) was designed primarily to overcome the shortcomings of exponential amplification approaches. TLAD uses a linear amplification method based on the in vitro transcription (IVT) of template DNA by RNA polymerase from the T7 phage, a common strategy used in RNA amplification protocols. The TLAD protocol introduced here successfully addresses amplification fidelity issues encountered with use of random PCR (R-PCR) with the ChIP-chip method (whereby DNA recovered from chromatin immunoprecipitation [ChIP] of cell lysate is used for subsequent analysis on DNA microarrays). Thus, optimizations have been done with this particular application in mind. For other applications that require DNA instead of RNA to be the end point, reverse transcription is a necessary step that increases the cost and complexity of necessary molecular biological manipulations to the sample, when compared with PCR. However, IVT amplification does offer improved fidelity and a much higher maximum yield per single reaction. Thus, other techniques that require amplification of complex mixtures of randomly fragmented genomic DNA can also benefit from this method.
RELATED INFORMATION
See the introduction by Marko et al. (2005) for a summary of the current state of the RNA amplification field. TLAD was originally described by Liu et al. (2003). The method is detailed (with modifications) in Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): I. CIP Treatment of Samples and Tailing Reaction with Terminal Transferase (Liu et al. 2008a), Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): II. Second-Strand Synthesis and In Vitro Transcription (Liu et al. 2008b), and Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): III. Sample Purification (Liu et al. 2008c).
BACKGROUND
Amplification of nucleic acids has become a mainstay of molecular biology. It permits genomic and transcriptional analysis when the amount of tissue or number of cells being studied become limiting and is invaluable in studies in which the biology of the specimens being investigated severely limits the amount of nucleic acids available. It is also particularly important for large-scale, high-throughput genomic studies, since these studies typically use microarrays or other high-throughput assays that often require microgram amounts of nucleic acids. Furthermore, these studies may use a large matrix of many different conditions and/or time points, which may make it prohibitively expensive to generate sufficient amounts of unamplified material.
One example of such a genomics application is the ChIP-chip method. This method (for review, see Buck and Lieb 2004) is typically used to identify transcription-factor binding sites and to map histone variants, histone post-translational modification patterns, or any other interesting epitope within the genome. Figure 1 shows an example of the ChIP-chip method using spotted microarrays. ChIP, however, typically yields DNA in the nanogram range, which is insufficient for most DNA microarray applications. Laboratories using the ChIP-chip technique typically employ an exponential amplification method, such as ligation-mediated PCR (LM-PCR) (Ren et al. 2000) or R-PCR (Bohlander et al. 1992; Gerton et al. 2000; Iyer et al. 2001), to obtain the quantities necessary for microarray analysis. R-PCR involves the annealing of primer adapters with a 5′ conserved end and a 3′ degenerate end to the template DNA, followed by extension and subsequent PCR amplification with primers complementary to the 5′ conserved ends. These exponential amplification methods adequately fulfill the needs of ChIP-chip analysis and are still frequently used, particularly when amplification from subnanogram amounts is desired. However, they have several shortcomings, in particular with regard to amplification fidelity and to a lesser extent with dynamic range compression, and with their inefficient amplification of short nucleic acids such as those <250 bp in length (Liu et al. 2003).
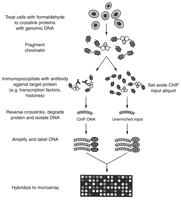
Schematic of the ChIP-chip method. Cells are first cross-linked with formaldehyde prior to lysis and DNA fragmentation. Following fragmentation, chromatin is incubated with antibodies and subsequently immunoprecipitated with Protein A or Protein G beads, which bind to the Fc segment of the antibodies. Following elution from the beads, reversal of cross-links, and proteinase K digestion, ChIP samples are typically phenol-chloroform extracted, ethanol precipitated, and then treated with RNase A to eliminate RNA that has carried over from the immunoprecipitation. The ChIP samples and the unenriched input material are then amplified and labeled with fluorescent dyes. The ChIP sample is subsequently hybridized, along with the unenriched input, on a spotted microarray. (Reprinted with permission, © 2005 Scion Publishing Ltd.)
GENERAL STRATEGY
The general strategy used by the TLAD method requires nanogram quantities of dsDNA as the starting material and generates microgram amounts of amplified RNA. An overall schematic diagram is shown in Figure 2. Briefly, the strategy is to add a 3′ conserved end to the template dsDNA, using terminal deoxynucleotidyl transferase (TdT) tailing, which permits the addition of a T7 promoter sequence in the subsequent second-strand synthesis step. IVT can then use this newly appended T7 promoter and linearly amplify the template dsDNA, producing amplified RNA product.
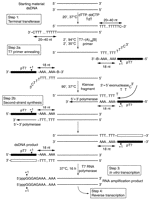
General strategy for the TLAD method. Starting with dsDNA template, TdT is used to add a poly(dT) tail to the 3′-ends of the template. This tail subsequently provides a conserved binding site for the annealing of T7 promoter (pT7)-poly(dA) primer adapters. Following subsequent second-strand synthesis using the large fragment of DNA polymerase I (Klenow fragment), one pair of dsDNA templates, with each pair member representing one of the two complementary strands of the dsDNA, is generated, with a T7 promoter at the 5′-end of the amplicon. In the subsequent IVT step, RNA is transcribed from this template in an isothermal reaction, producing an RNA amplification product consisting of both strands of the original dsDNA template in high microgram quantities. Note that each RNA strand will contain a short sequence from the T7 promoter and a poly(A) tract, 5′ relative to the amplicon. (Reprinted with permission from Liu et al. [2003].)
CONSIDERATIONS FOR THE STARTING dsDNA TEMPLATE
The ideal starting material for this method is dsDNA template in the 100- to 1000-bp size range with 3′ protruding or 3′ blunt ends. The 3′-end must have a free hydroxyl (OH) group, since TdT does not add residues to template strands with 3′ phosphate groups. Some restriction digests and other DNA fragmentation methods such as sonication and nuclease digestion may leave behind 3′ phosphate groups on a significant proportion of the DNA molecules. These phosphate groups need to be removed prior to the tailing reaction; see below, and Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): I. CIP Treatment of Samples and Tailing Reaction with Terminal Transferase (Liu et al. 2008a). Failure to do so may result in poor amplification.
One other consideration about 3′-ends is their efficacy in serving as an efficient template for TdT-mediated polynucleotide tailing. The efficiency of the TdT enzyme is maximal on 3′ protruding ends and good on 3′ blunt ends. However, 3′ recessed ends are only “very reluctantly” tailed, and a mixture of templates containing all three types of 3′-end will likely result in tailed products enriched with previously 3′ protruding ends and depleted of previously 3′ recessed ends (Sambrook and Russell 2001). The end result is a larger spread in the size distribution of poly(dT) tail lengths, which may have a negative impact on subsequent amplification fidelity if the relationship between the template sequence and the nature of the 3′-end is nonrandom. It is therefore recommended that, in applications that are sensitive to this issue, templates containing a significant proportion of 3′ recessed ends are filled in with Klenow fragment DNA polymerase prior to the TdT tailing step.
The size range of ~100-1000 bp is considered to be optimal, as this is the size range that was extensively tested with the TLAD method when it was developed. Amplification of fragments smaller than 100 bp is limited by the column-based purification steps, which rely on QIAGEN MinElute columns (see Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): I. CIP Treatment of Samples and Tailing Reaction with Terminal Transferase [Liu et al. 2008a] and Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): II. Second-Strand Synthesis and In Vitro Transcription [Liu et al. 2008b]) and RNeasy columns (see Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): III. Sample Purification [Liu et al. 2008c]). The MinElute columns have a lower limit of 70 bp, while the RNeasy columns have a tested limit of 100 bp (which is lower than the 200-bp manufacturer-specified limit). Although this method was developed with QIAGEN columns, alternative reaction clean-up columns or other methods can be substituted.
Amplification of material smaller than 100 bp will also be less efficient in terms of mass. This is because the rate-limiting step in IVT is initiation of transcription (Martin et al. 1988), and because there is a greater molar ratio of template molecules to T7 RNA polymerase compared with an equivalent mass of a higher-molecular-weight template. Here, the low-molecular-weight template would require more initiation of transcription events per mass unit compared with a high-molecular-weight template, and the usual result is a significantly lower mass yield for the low-molecular-weight template.
Amplification of fragments larger than 1000 bp has not been extensively tested. This should not be a particular problem for templates ranging up to 4-5 kb in size, since T7 RNA polymerase is highly processive (Martin et al. 1988). Extremely large fragments (>5 kb) are perhaps best amplified by other techniques (e.g., strand displacement) (Lage et al. 2003).
USING THIS METHOD FOR ChIP-chip EXPERIMENTS
Because this method was originally designed to address the amplification needs of ChIP-chip experiments, considerations necessary to optimize amplification fidelity and yield of starting material are detailed here. However, this approach will be appropriate for many applications that require several micrograms of DNA from nanogram quantities of starting DNA. Additional information is provided in the Troubleshooting section of Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): III. Sample Purification (Liu et al. 2008c).
ChIPs frequently yield low amounts of DNA, which may be difficult to quantify via usual methods, such as absorbance at 260 nm with a spectrophotometer. Most commonly used spectrophotometers have a lower reliable detection limit of 0.01 absorbance units and are limited to a minimum cuvette volume of 50-100 μL, translating to 0.5 ng/μL for dsDNA. Consequently, it becomes quite difficult to detect and quantify reliably the presence of nucleic acid when the total yield is <250 ng, without sacrificing an excessive proportion of the total sample in the measurement. Reliable measurement of a ChIP mass yield is important for maximizing amplification yield for this method. Therefore, we recommend using a more sensitive instrument or a spectrophotometer designed to handle cuvettes with small volumes (<10 μL). Since these instruments require fivefold to 10-fold less sample per measurement, the lower limit then becomes 25-50 ng of ChIP yield for reliable measurement. For extremely low ChIP yields (<25 ng), quantification via fluorescence (e.g., using PicoGreen [Molecular Probes] with the Turner BioSystems TD-700 fluorometer) is the recommended method and should reliably measure samples down to a lower limit of 2.5 ng of starting material.
There are additional technical considerations when TLAD is used with the ChIP-chip method. Calf intestinal alkaline phosphatase (CIP) treatment is presented as an optional protocol (see Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): I. CIP Treatment of Samples and Tailing Reaction with Terminal Transferase [Liu et al. 2008a]), but is necessary if the ChIP-chip method uses fragmentation methods (e.g., sonication or nuclease digestion) that leave behind a significant proportion of 3′ phosphate groups in the template DNA. Additionally, since CIP treatment is typically carried out immediately after RNase A digestion in the ChIP-chip method, users may be concerned about sufficient elimination of RNase A for the remainder of the method, in time for the IVT step (see Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): II. Second-Strand Synthesis and In Vitro Transcription [Liu et al. 2008b]). The three MinElute clean-up steps (performed after CIP treatment, TdT tailing, and second-strand synthesis) are usually sufficient to remove enough residual RNase A for it not to pose a problem. Furthermore, the Ambion IVT enzyme mix used in the protocol contains an RNase inhibitor. Care should be taken, however, to ensure that barrier tips are used, and ideally separate pipettes should be used when handling the RNase A enzyme to avoid RNase contamination.
CONTROLS FOR NEW USERS
To maximize the success of this method, two positive controls are strongly suggested for new users. In addition, the Troubleshooting section of Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): III. Sample Purification (Liu et al. 2008c) discusses in greater detail several strategies that can be used to resolve low-yield problems.
Positive Amplification Control
Include this control during Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): I. CIP Treatment of Samples and Tailing Reaction with Terminal Transferase (Liu et al. 2008a) and Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): II. Second-Strand Synthesis and In Vitro Transcription (Liu et al. 2008b). This control enables the user to distinguish sample-specific problems from protocol-implementation issues. Here, the starting material is 50 μg of blunt-ended PCR product in the 100- to 1000-bp range (preferably around 200-500 bp). If this control is used as part of a ChIP-chip experiment, the PCR product can be subject to RNase A and CIP treatment for troubleshooting purposes, although in practice these treatments are usually not necessary. Yields should typically range from 30 to 60 μg (with a maximum of 80-100 μg observed), depending on the size of the PCR product, protocol implementation, and quality of the reagents used (particularly the nucleotides used for IVT, which are highly sensitive to freeze-thaw cycles).
Positive IVT Control
Include this control during Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): II. Second-Strand Synthesis and In Vitro Transcription (Liu et al. 2008b). This control enables the user to distinguish sample-specific problems from IVT-specific problems (such as RNase contamination). Here, the IVT starting material is 250 ng of the pTRI-Xef linearized plasmid provided with the Ambion IVT kit. If not using the kit, an appropriate amount of a dsDNA template that contains the pT7 promoter, with a prior history of use as a successful T7 RNA polymerase template, should be used. Yields should typically range from 100 to 140 μg, limited by exhaustion of the nucleotides in the reaction mixture and the rated 100-μg binding capacity of the QIAGEN RNeasy column. Any yield somewhat less than this may suggest RNase contamination, evaporation of the reaction mixture during the long incubation period (due to the small reaction volume), or poor-quality reagents (particularly the nucleotides). Visualization on a nondenaturing agarose gel should yield two intense bands of ~0.9 and 1.5 kb, as shown in Figure 3 (lane 2).
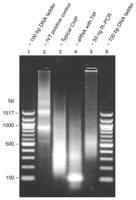
Amplification products on a nondenaturing 2% agarose gel in TAE buffer, stained with ethidium bromide. A 100-bp ladder (500 ng; New England Biolabs) or 650 ng of samples were loaded. (Lanes 1,6) 100-bp ladder; (lanes 2-4) aRNA amplified using the method presented here, with the following templates: (lane 2) IVT positive control (pTRI-Xef); (lane 3) a typical ChIP sample (S. cerevisiae bis-acetyl histone H3 on lysine 9 and 14) from sonicated genomic DNA; (lane 4) amplification from 5 ng of a genomic digest of S. cerevisiae with RsaI, showing a strong band of template-independent product (TIP) of ~100 bp; (lane 5) the R-PCR product, for comparison, from a 50-ng amplification of the same template used to generate aRNA in lane 4. The most remarkable attribute of lane 5 is the near absence of DNA smaller than 250 bp. (Reprinted with permission, © 2005 Scion Publishing Ltd.)
EXPECTED YIELDS
Depending on the amount and size distribution of the starting material, the typical yield is 5-90 μg of antisense RNA (aRNA), as detailed in Table 1. The amplification yield for a given starting amount is represented by a range, which is dependent on the quality and size distribution of the starting material. A more direct determinant of amplification yield will be based on the number of picomoles of template used. Initiation of the reaction is the rate-limiting step for TdT tailing and IVT, thus the number of template molecules present will more directly determine the actual amplification yield. For example, amplifying 50 ng of a 200-bp PCR product is likely to produce a lower mass yield than amplifying 50 ng of a 800-bp PCR product, even though the number of aRNA molecules produced may actually be greater for the 200-bp product than for the 800-bp product. For comparison, the R-PCR yield (30 cycles) of a 50-ng starting amount is shown in Figure 3 (lane 5).
COMPOSITION OF THE AMPLIFICATION PRODUCT
Typically, a 1%-2% agarose gel, containing or stained with ethidium bromide, is used to assess the composition and quality of the amplified RNA (Fig. 3). Unless size distribution is crucial, it is usually not necessary for the gel to be denaturing. Within the resolution limits of the agarose gel, the amplified product may shift higher on the gel, in the order of 20-40 bp. This shift is to be expected because of the addition and tightness of the size distribution of the poly(A) tail, plus the 5-nucleotide (nt) sequence added by the T7 promoter. The size distribution of the poly(A) tail becomes particularly evident in amplification products produced from a single-size template, such as PCR products or a restriction-digested plasmid. Occasionally, a substantial amount of a low-molecular-weight band may also appear near the bottom of the gel, at ~100 bp (see Fig. 3, lane 4). This band has been observed under certain amplification conditions, usually when the concentration of starting material is significantly less than that of the primer during second-strand synthesis. Although the composition of this band has not been tested, it likely represents the amplification product produced from IVT-valid template synthesized through the formation of primer dimers during second-strand synthesis. This side reaction consumes reagents and activity of the RNA polymerase enzyme, reducing the true effective yield of the desired amplification product. To minimize this side reaction, established optimum primer-to-template mass ratios (roughly 5:1) are included in Table 1 of Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): II. Second-Strand Synthesis and In Vitro Transcription (Liu et al. 2008b).
As mentioned above, the QIAGEN MinElute kit can be used a number of times to remove buffer salts, spent enzymes, free nucleotides, and any DNA products shorter than 40 nt. Material between 40 and 70 nt may also be removed, but at a lower efficiency.
RESULTS
Summary of Amplification Fidelity
A detailed discussion of the validation procedure for amplification fidelity and reproducibility will not be repeated here, since it has been previously described in detail (Liu et al. 2003). However, key points from this discussion will be briefly mentioned. First, within the 100- to 700-bp range tested for this method, the size distribution of the amplification product is effectively preserved, compared with R-PCR, which loses material below 250 bp and subsequently has reduced fidelity (see Fig. 3, lane 5). IVT amplification also produces an increased dynamic range that does not appear to have a negative impact on its fidelity, improving the prospects of separating biological trends from the noise inherent in microarray experiments. This is another improvement over R-PCR, which often suffers from dynamic range compression. Furthermore, IVT amplification retains fidelity and representation of the starting material when compared with an unamplified Klenow-labeled standard.
Updates to the Method
Since the initial publication of this method (Liu et al. 2003), several updates have been incorporated. Although most of the changes are minor, a number of more significant changes have been implemented. The protocol for Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): II. Second-Strand Synthesis and In Vitro Transcription (Liu et al. 2008b) was optimized with the New England Biolabs (NEB) Klenow enzyme for the second-strand synthesis step. Recently, NEB switched from supplying EcoPol buffer with this enzyme to supplying NEB Buffer 2. This switch has now been tested, with results indicating no change or a slight improvement (5%-15%) of yields. Furthermore, the second-strand synthesis step now contains a detailed table of optimized primer amounts and recommended final reaction volumes for a given starting amount; see Table 1 of Whole Genome Amplification by T7-Based Linear Amplification of DNA (TLAD): II. Second-Strand Synthesis and In Vitro Transcription (Liu et al. 2008b). These measures combine to reduce the side reaction of low-molecular-weight material generated from speculated primer-dimer formation from excess primer (Baugh et al. 2001; Liu et al. 2003). Moreover, the default reaction volume has been reduced from 50 to 25 μL for the recommended starting amount of 50-75 ng, which permits the reduction of reagent costs without affecting yield. Finally, for the IVT step, amplification of low-molecular-weight material (i.e., <300 bp) has been optimized by boosting the amount of T7 enzyme used per reaction. The boost typically results in a 10%-30% yield improvement, likely due to a more favorable stoichiometric ratio of the T7 enzyme to the DNA template.
ChIP-chip RESULTS
The method presented here has already been used in several published studies (Bernstein et al. 2004, 2005; Humphrey et al. 2004; Lee et al. 2004). An example of the results obtained from the ChIP-chip method, when used in conjunction with TLAD, is shown in Figure 4. Here, the ChIP-chip method was performed on Saccharomyces cerevisiae histone H3 and Flag-H2B, carried through the DNA linear amplification method presented here, reverse-transcribed, labeled, and hybridized onto S. cerevisiae genome arrays consisting of intergenic regions. The enrichment ratio was plotted against the transcription rate of downstream genes; one can observe the inverse relationship between nucleosome occupancy and promoter strength.
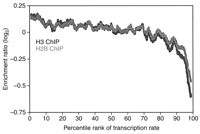
Example of ChIP-chip results. Results from ChIPs of S. cerevisiae histone H3 and Flag-H2B, plotted by intergenic genomic loci as a function of the percentile rank of the transcription rate in the downstream genes (Bernstein et al. 2004). An inverse relationship can be seen between nucleosome occupancy (as represented by enrichment for histone H3 and H2B) and promoter strength, as indicated by the transcription rate. These ChIP samples were amplified with the DNA linear amplification protocol introduced here. (Reprinted with permission from Bernstein et al. [2004].)
ACKNOWLEDGMENTS
C.L.L. is supported by a Graduate Research Fellowship from the National Science Foundation. S.L.S. is an investigator at the Howard Hughes Medical Institute. B.E.B. is supported by a K08 Development Award from the National Cancer Institute. This work was supported by a grant from the National Institute for General Medical Sciences.








