
Preparation of Small RNA Libraries for High-Throughput Sequencing
Adapted from RNA: A Laboratory Manual, by Donald C. Rio, Manuel Ares Jr, Gregory J. Hannon, and Timothy W. Nilsen. CSHL Press, Cold Spring Harbor, NY, USA, 2011.Abstract
This protocol details the process of small RNA cloning for sequencing on the Illumina/Solexa sequencing platform, but it can be easily modified for use on other next-generation platforms (e.g., SOLiD, 454). This procedure is designed to clone canonical small RNA molecules with 5′-monophosphate and 3′-hydroxyl termini. Modifications, such as the presence of a 2′-O-methyl group, can reduce efficiency, although not sufficiently to negate the utility of the approach. Other termini modifications, such as a 5′ triphosphate or a 3′ phosphate, can be altered by enzymatic treatment before cloning.
MATERIALS
Reagents
Bovine serum albumin (BSA; 100×)
Chloroform
Decade Marker (Ambion)
Diethyl pyrocarbonate (DEPC)–Milli-Q H2O
Dimethylsulfoxide (DMSO)
Ethanol (100%, 70%)
Ethidium bromide
[γ-32P]ATP (Perkin Elmer)
Gel Loading Buffer II (1×) (Ambion)
Gel-loading dye (6×) (Fisher)
GeneRuler 50-bp DNA ladder (Crystalgen)
GlycoBlue (Ambion)
NaCl (0.4 m)
Oligonucleotides (custom-prepared, HPLC-purified):
-
BanOne: 5′-ATTGATGGTGCCTACAG-3′ (3′ primer for reverse transcription)
-
DNA oligonucleotide of appropriate length that contains a PmeI restriction site (see Step 2)
-
Modban: AMP-5′p = 5′-pCTGTAGGCACCATCAATdideoxyC-3′ (miRNA cloning linker 1 [IDT]) (for 3’ ligation)
-
Sol_3_Modban: 5′-CAAGCAGAAGACGGCATACGATTGATGGTGCCTACAG-3′ (for PCR)
-
Sol_5_SBS3: 5′-AATGATACGGCGACCACCGAACACTCTTTCCCTACACGACG-3′ (for PCR)
-
Solexa linker: 5′-rArCrArCrUrCrUrUrUrCrCrCrUrArCrArCrGrArCrGrCrUrCrUrUrCrCrGrArUrC-3′ (for 5′ ligation)
PCR buffer (Roche)
Phenol (equilibrated, pH 8)
Phenol:chloroform:isoamyl alcohol (PCA)
PmeI (New England BioLabs)
-
This includes 10× buffer and BSA.
PNK buffer (Roche)
Polyacrylamide/urea gel (Sequagel, National Diagnostics)
Reverse transcriptase mix stock
Sample of RNAs to be cloned
Seakem GTG agarose (low melting, <1-kb DNA/RNA)
SuperScript III RT (Invitrogen)
-
This includes 5× buffer, DTT, and dNTP mix.
SYBR Gold nucleic acid stain (Molecular Probes)
T4 polynucleotide kinase (New England Biolabs)
-
This includes 10× buffer.
T4 RNA ligase (Ambion)
-
This includes 10× buffer.
T4 RNA ligase 2 (truncated; New England Biolabs)
-
This includes 10× buffer.
TAE
Taq polymerase (Roche)
-
This includes 10× buffer + MgCl2 and dNTP mix.
TBE (1×) (for extraction of RNA by dialysis; see Step 24)
Equipment
Chemical fume hood
Dialyzer tube (Novagen Dialyzer Midi, MWCO 3.5 kD, 71506-3) (for extraction of RNA by dialysis; see Step 24)
Ethanol (100%)-dry ice bath (in an insulated container)
Gel box for dialysis (for extraction of RNA by dialysis; see Step 24)
Gel electrophoresis equipment
Microcentrifuge tubes (low retention; Axygen MAXYMum Recovery microtubes)
Microcentrifuge tubes with micropore filter (Ultrafree-MC, 0.22 µm; Millipore)
Paper triangles, small (to serve as reference markers; see Step 7)
Pestle
Phosphorimaging screen
Pipettor with a fine, RNase-free tip
Razor blade
Sepharose column (G-50; GE Healthcare Life Sciences)
Thermal cycler
Ultraviolet (UV) light (long wave)
Vortex mixer
METHOD
The general procedure and 3′ cloning linker were adapted from Lau et al. (2001). RNA samples are sensitive to degradation at all steps of the procedure; use RNase-free reagents and supplies when able to do so. Gels, samples, and supplies will contain radioactivity, so always dispose of materials properly and follow proper radiation safety procedures.
Section I: Size Selection and Gel Purification of RNA Sample
-
1. Prepare a 15% vertical polyacrylamide/urea gel.
-
The thickness of the gel depends on the amount of total RNA being run. As a guide, we use 1.0 mm for <20 µg and 1.5 mm for >20 µg.
-
-
2. Prepare the following 5′ oligonucleotide-labeling reaction for 19-, 24-, 28-, and/or 32-nucleotide RNA oliogonucleotides (depending on the small RNA sizes to be cloned) and incubate for 30 min at 37°C:
-
2 µL PNK buffer (10×)
-
1 µL DNA oligonucleotide (10 µm)
-
2–5 µL [γ-32P]ATP
-
1 µL T4 polynucleotide kinase
-
H2O to 20 µL
Eliminate free ATP by running the samples through a G-50 Sepharose column.
-
Oligonucleotides must contain an internal PmeI recognition sequence.
-
-
3. Load 10 ng of 32P-labeled Decade Marker as a size marker.
-
4. Add an equal amount of Gel Loading Buffer II to the mixture. Heat the samples for 5 min at 95°C and then chill for 1 min on ice.
-
5. Spike the RNA samples with 32P-labeled oligonucleotides (∼10,000 counts/oligonucleotide per min).
-
6. Load the sample(s) on a 15% polyacrylamide gel and run the gel at a constant 10 W for 1–1.5 h (until the first dye front reaches the middle of the gel).
-
In addition, 1–5 µg of each RNA sample can be loaded on a separate 1.0-mm gel, run, stained with SYBR Gold, and visualized under a UV lamp to verify RNA integrity.
-
-
7. Once the run has completed, pull one glass plate away from the gel and place it face up behind a clear radioactive shield. Then, blot a tiny amount of radioactivity onto small cut-out paper triangles and place them at three points on the face of the gel.
-
These will serve as references when lining up the gel on the radioactive image where the bands should be cut out.
-
-
8. Expose a phosphor screen over the gel for 10–15 min (time will vary depending on radioactivity intensity and imager sensitivity) (Fig. 1). Print the gel image at actual size so that it can be placed under the glass plate and gel to use as a reference to cut out small RNAs of desired size in the following step.
-
9. Excise the precise band from the gel that corresponds to the desired size of small RNA (including radiolabeled oligonucleotide[s]) into a low-retention microcentrifuge tube (use these throughout the protocol to minimize sample loss).
-
10. Reexpose the gel under a phosphor screen to ensure that the desired small RNA fragments have been extracted from the gel.
-
The selection of 20–28-nucleotide-sized small RNAs is shown in Figure 1. Follow Steps 11–23 for grinding the slices and extracting by diffusion. For a higher yield of RNA from polyacrylamide gel slices, go to Step 24 to perform dialysis extraction.
-

Size selection and gel extraction of small RNAs from total RNA. Radioactively labeled markers and oligonucleotides are resolved with unlabeled total RNA on a 12% polyacrylamide gel; 24- and 28-nucleotide markers were run in a separate lane to allow for more accurate size resolution. Total RNA samples were spiked with a radiolabeled 24-nucleotide oligonucleotide (although any combination of labeled oligonucleotides can be used), allowing one to visually follow the ligation process and also ensuring that the desired size fraction has been extracted from the gel. Here, 20–28-nucleotide small RNAs (including the radiolabeled 24-nucleotide oligonucleotide) have been cut from the gel. The gel was then reexposed, showing the removal of radiolabeled oligonucleotides (and underlying small RNAs).
Extraction by Diffusion
-
11. Centrifuge the excised gel slice briefly in a centrifuge at maximum speed for 1 min at room temperature.
-
12. Carefully grind gel slices by hand using a pestle.
-
13. Centrifuge the microcentrifuge tube for 1 min at room temperature to pellet the ground gel to the bottom of the tube.
-
14. Add 420 µL of 0.4 m NaCl to the ground gel slices.
-
15. Flash-freeze the samples for 1 min in a bath of ethanol and dry ice.
-
16. Incubate overnight at room temperature with agitation (secure the samples on a vortex).
-
17. Centrifuge the gel slice homogenate through a micropore filter at full speed for 1 min at room temperature.
-
18. Transfer the filtrate to a fresh microcentrifuge tube.
-
19. Add 20 µg of GlycoBlue, vortex to mix, and add 2.5 volumes of 100% ethanol.
-
20. Incubate the mixture for 3–6+ h at –20°C.
-
21. Centrifuge at full speed for 30 min at 4°C.
-
22. Wash with 70% ethanol and then remove all of the ethanol using a pipettor with a fine, RNase-free tip.
-
23. Air-dry the pellet for <5 min and resuspend it in 13 µL of DEPC-Milli-Q H2O.
-
Small RNAs of the desired size range have now been purified. Proceed to Step 25.
-
Extraction by Dialysis
-
24. Alternatively, extract the sample from the gel using a gel box and dialyzer.
-
i. Insert a gel slice into a dialyzer tube with 450 µL of H2O.
-
ii. Position the dialyzer into a gel box, submerged in 1× TBE, such that the dialysis tubing has the current running perpendicularly through it.
-
iii. Run at 100 V for 18 min.
-
iv. Reverse the poles and run for 2 min to pull the sample off the dialysis tubing wall.
-
v. Pull out all of the water into a fresh microcentrifuge tube, mix in 20 µg of GlycoBlue, bring the solution to 0.4 m NaCl, mix, and add 2.5 volumes of 100% ethanol.
-
vi. Mix by inverting and incubate for 3–6+ h at –20°C.
-
vii. Centrifuge at full speed for 30 min at 4°C.
-
viii. Wash the pellet with 70% ethanol and then remove all of the ethanol.
-
ix. Air-dry the pellet for <5 min and resuspend it in 13 µL of DEPC-Milli-Q H2O.
-
Small RNAs of the desired size range have now been purified.
-
-
Section II: 3′ Linker Ligation
-
25. Set up the following reaction (20 µL total volume) in a microcentrifuge tube:
-
13 µL gel-purified RNA (in H2O) (from Step 23 or 24)
-
2 µL ATP-free T4 RNA ligase buffer (10×)
-
2 µL DMSO
-
1 µL Modban oligonucleotide (50 µm)
-
2 µL T4 RNA ligase 2, truncated
25. Incubate for 1 h at 37°C.
-
-
26. Add 20 µL of Gel Loading Buffer II and then heat-inactivate for 5 min at 95°C.
Section III: Gel Purification of 3′-ligated RNA Product
-
27. Heat samples for 5 min at 95°C and then chill on ice for 1 min.
-
28. Load 5 ng of 32P-labeled Decade Marker as a size marker.
-
29. Prepare a 1.0-mm 15% vertical polyacrylamide/urea gel.
-
30. Load the sample(s) and run the gel at a constant 10W for 1.5–2 h (until the first dye front reaches the bottom of the gel).
-
31. Once the run has completed, pull one glass plate away from the gel and place it face up. Then, blot a tiny amount of radioactivity onto small cut-out paper triangles and place them at three points on the face of the gel. These will serve as references when lining up the gel on the radioactive image and align where the bands should be cut out.
-
32. Expose a phosphorimager screen over the gel for 30–45 min (time will vary). Print the gel image at actual size so that it can be placed under the glass plate and gel to use as a reference to cut out small RNAs of desired size in the following step.
-
33. Excise the precise band corresponding to the desired size of small RNA (including the ligated radiolabeled oligonucleotide[s]) into a microcentrifuge tube.
-
Selection of 38–47-nucleotide-sized small RNAs is shown in Figure 2. The steps below describe how to grind the gel slices and extract the RNA by diffusion. For a higher yield of RNA, perform dialysis extraction as described in Step 24 above.
-
-
34. Centrifuge the gel slice briefly at maximum speed for 1 min at room temperature.
-
35. Carefully grind the gel slices using a pestle.
-
36. Centrifuge the microcentrifuge tube for 1 min at room temperature to pellet the ground gel to the bottom of the tube.
-
37. Add 420 µL of 0.4 m NaCl to the ground gel slice.
-
38. Quickly freeze the samples for 1 min in a bath of ethanol and dry ice.
-
39. Incubate overnight at room temperature with agitation (secure the sample on a vortex).
-
40. Centrifuge the gel slice homogenate through a micropore filter at full speed for 1 min at room temperature.
-
41. Transfer the filtrate to a microcentrifuge tube.
-
42. Add 20 µg of GlycoBlue, mix, and add 2.5 volumes of 100% ethanol.
-
43. Incubate the mixture for 3–6+ h at –20°C.
-
44. Centrifuge at full speed for 30 min at 4°C.
-
45. Wash the pellet with 70% ethanol and then remove all of the ethanol.
-
46. Air-dry the pellet for <5 min and then resuspend it in 13 µL of DEPC-Milli-Q H2O.
-
3′-end cloned small RNA molecules have now been purified.
-
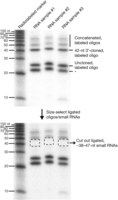
Size selection and gel extraction of 3′-ligated small RNAs. Radioactively labeled markers and ligated oligonucleotides were resolved on a 10% polyacrylamide gel. The 20–28-nucleotide small RNAs (including the 24-nucleotide radiolabeled oligonucleotide) now have an 18-nucleotide linker ligated to their 3′ termini, leading to a predicted size shift of 18 nucleotides. Here, ∼38–47-nucleotide small RNAs (including the ligated, radiolabeled 24-nucleotide oligonucleotide, now running at 42 nucleotides) have been cut from the gel. The gel was then reexposed, showing the removal of radiolabeled oligonucleotides (and underlying small RNAs). Although we have not characterized additional products on the gel, based on size, we have indicated the likely origin of labeled species. Asterisk indicates bands of unknown production.
Section IV: 5′ Linker Ligation
-
47. Set up the following reaction (20 µL total volume):
-
13 µL ligated RNA product (in H2O)
-
2 µL T4 RNA ligase buffer (10×)
-
2 µL DMSO
-
1 µL Solexa linker (50 µm)
-
2 µL T4 RNA ligase
Incubate for 1 h at 37°C.
-
48. Add 20 µL of Gel Loading Buffer II and then heat-inactivate for 5 min at 95°C.
-
Section V: Gel Purification of 5’- and 3’-ligated RNA Product
-
49. Heat the samples from Step 48 for 5 min at 95°C and then chill for 1 min.
-
50. Load 2.5 ng of 32P-labeled Decade Marker as a size marker.
-
51. Prepare a 1.0-mm 15% vertical polyacrylamide/urea gel.
-
52. Load the sample(s) and run the gel at a constant 10 W for 2+ h (until the first dye front passes the bottom of the gel).
-
53. Once the run has completed, pull one glass plate away from the gel and place it face up. Then, blot a tiny amount of radioactivity onto small cut-out paper triangles and place them at three points on the face of the gel. These will serve as references when lining up the gel on the radioactive image and align where the bands should be cut out.
-
54. Expose a phosphorimager screen over the gel for 1+ h (time will vary). Print the gel image at actual size so that it can be placed under the glass plate and gel to use as a reference to cut out small RNAs of desired size in the following step.
-
55. Excise the precise band corresponding to the desired size of small RNAs (including the ligated radiolabeled oligonucleotide[s]) into a microcentrifuge tube.
-
Selection of 69–79-nucleotide-sized small RNAs is shown in Figure 3. The steps below describe how to grind the gel slices and extract the RNA by diffusion. For a higher yield of RNA, perform dialysis extraction as described in Step 24 above.
-
-
56. Centrifuge the gel slice briefly at maximum speed for 1 min at room temperature.
-
57. Carefully grind the gel slices using a pestle.
-
58. Centrifuge the microcentrifuge tube for 1 min at room temperature to pellet the ground gel to the bottom of the tube.
-
59. Add 420 µL of 0.4 m NaCl to the sample.
-
60. Quickly freeze the samples in a bath of ethanol and dry ice for 1 min.
-
61. Incubate overnight at room temperature with agitation (secure the sample on a vortex).
-
62. Centrifuge the gel slice homogenate through a micropore filter at full speed for 1 min at room temperature.
-
63. Transfer the eluant filtrate to a microcentrifuge tube.
-
64. Add 20 µg of GlycoBlue, mix, and add 2.5 volumes of 100% ethanol.
-
65. Incubate the mixture for 3–6+ h at –20°C.
-
66. Centrifuge for 30 min at 4°C.
-
67. Wash the pellet with 70% ethanol and then remove all of the ethanol.
-
68. Air-dry for <5 min and resuspend the pellet in 6.3 µL of DEPC-Milli-Q H2O.
-
3′- and 5′-end cloned small RNA molecules have now been purified.
-

Size selection and gel extraction of 3′- and 5′-ligated small RNAs. Radioactively labeled markers and ligated oligonucleotides are resolved on a 10% polyacrylamide gel. The ∼38–47-nucleotide small RNAs (including the 42-nucleotide radiolabeled oligonucleotide) now have a 32-nucleotide linker ligated to their 5′ termini, leading to a predicted size shift of 32 nucleotides. Here, ∼69–79-nucleotide small RNAs (including the ligated, radiolabeled 24-nucleotide oligonucleotide, now running at 74 nucleotides) have been cut from the gel. The gel was then reexposed, showing the removal of radiolabeled oligonucleotides (and underlying small RNAs). Although we have not characterized additional products on the gel, based on size, we have indicated the likely origin of labeled species. Asterisk indicates bands of unknown production.
Section VI: Reverse Transcription
-
69. Set up the following reaction in a 150-µL PCR thermocycler tube and incubate for 2 min at 72°C.
-
6.3 µL ligated RNA product (in H2O)
-
4.2 µL BanOne primer (5 µm)
-
-
70. Centrifuge for 1 min at room temperature.
-
71. Cool for 2 min on ice.
-
72. Add 8.4 µL of reverse transcriptase mix stock.
-
73. Split the mixture into two tubes (9 µL each).
-
74. Add either 1 µL SuperScript III RT (Invitrogen) (+RT) or 1 µL DEPC–Milli-Q H2O (–RT control).
-
75. Incubate for 1 h at 50°C and then heat for 15 min to 70°C.
-
76. Store at –20°C until use or proceed directly to Section VII.
Section VII: PCR Amplification of cDNA
-
77. Set up two PCRs (100 µL total volume each) for each sample (+RT and –RT) in 150-µL PCR thermocycler tubes.
-
5 µL first-strand cDNA (+RT) or control (–RT) (from Step 76)
-
10 µL PCR buffer + MgCl2 (10×)
-
2 µL dNTP mix (10 mm of each)
-
1 µL Sol_5_SBS3 (100 µm)
-
1 µL Sol_3_Modban (100 µm)
-
80 µL Milli-Q H2O
-
1 µL Taq polymerase (5 units/µL) (high-fidelity polymerase can be used)
-
-
78. Place the reaction in a thermal cycler and run the following PCR program:
-
5 cycles: 94°C for 2 min, 94°C for 15 sec, 54°C for 30 sec, 72°C for 30 sec
-
15–25 cycles: 94°C for 15 sec, 60°C for 30 sec, 72°C for 30 sec
-
The cycle range depends on the amount of starting RNA, small RNA abundance, and ligation/elution efficiencies; we recommend as few repeats as possible.
-
-
72°C for 2 min, 4°C hold
-
-
79. Transfer the +RT samples to fresh microcentrifuge tubes and add 200 µL of 0.5 m NaCl.
-
80. Add 300 µL of PCA and vortex for 1 min.
-
81. Centrifuge at maximum speed for 5 min at room temperature.
-
82. Transfer the aqueous layer to 900 µL (or 2.5–3 volumes) of 100% ethanol and 20 µg of GlycoBlue (optional).
-
83. Incubate the mixture for 3–6+ h at –20°C.
-
84. Centrifuge at maximum speed for 30 min at 4°C.
-
85. Wash the pellet with 70% ethanol and then remove all of the ethanol.
-
86. Wash the pellet with 100% ethanol and then remove all of the ethanol.
-
87. Air-dry the pellet and then resuspend it in 23.7 µL of H2O.
-
Your cDNA-converted small RNA library is now amplified.
-
Section VIII: PmeI Digestion of Radiolabeled Oligonucleotides
-
88. Set up the following reaction (30 µL total) and incubate for 2–4 h at 37°C:
-
23.7 µL PCR products (in H2O) from Step 87
-
3 µL NEB buffer 4 (10×)
-
3 µL PmeI
-
0.3 µL BSA (100×)
-
-
89. Add 6 µL of fresh 6× gel-loading dye.
-
All RNA size markers containing a PmeI restriction enzyme site are now cleaved away.
-
-
90. Proceed to Section IX.
Section IX: Gel Purification of Amplified cDNA
-
91. Prepare a 2% low-melt agarose gel (using Seakem GTG agarose) in TAE, according to the manufacturer’s instructions, including a minimal amount of ethidium bromide.
-
92. Load 8–12 µL of 50-bp DNA ladder, PCR-amplified library from cDNA (+RT), and control (–RT). Run the gel at a constant 80 V until the bromophenol blue band is two-thirds through the gel.
-
93. View the PCR products with a long-wave UV source (Fig. 4).
-
To maintain the integrity of the libraries, take all precautions to minimize the amount of time that the samples are exposed to UV light.
-
-
94. Excise the DNA band with a clean razor blade and transfer the gel slice into a preweighed microcentrifuge tube.
-
95. Centrifuge at full speed for 1 min at room temperature.
-
96. Add 0.5 m NaCl to a total volume of 500 µL.
-
97. Melt the agarose in the solution for 10 min at 70°C, flicking tube the every couple of minutes.
-
98. Place microcentrifuge tubes with 500 µL of phenol (equilibrated, pH 8) at 70°C.
-
99. Add hot phenol to the tubes with melted agar and vortex immediately for 1 min.
-
Perform under a chemical fume hood.
-
-
100. Centrifuge the mixture at maximum speed for 5 min at room temperature.
-
101. Transfer the aqueous layer to 1 volume of PCA, vortex, and centrifuge at maximum speed for 5 min at room temperature.
-
102. Transfer the aqueous layer to 1 volume of chloroform, vortex, and centrifuge at maximum speed for 5 min at room temperature.
-
103. Transfer the aqueous layer to 2.5–3 volumes of 100% ethanol.
-
104. Incubate for 2 hr to overnight at –20°C.
-
105. Centrifuge at maximum speed for 15 min at 4°C.
-
106. Wash the pellet with 70% ethanol and then remove all of the ethanol.
-
107. Wash pellet with 100% ethanol and then remove all of the ethanol.
-
108. Air-dry the pellet and then resuspend it in 20 µL of H2O.
-
109. Determine sample concentration and dilute to 10 nm before sequencing.
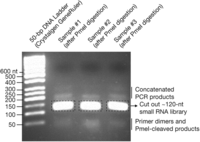
Size selection and gel extraction of small RNA libraries. Processed libraries are resolved by UV light on a 2% low-melting agarose gel stained with ethidium bromide. A 50-bp size marker and PmeI-digested PCR products of each library are shown. A primary band at ∼120 nucleotides is indicated, which was carefully cut and extracted from the gel (see Section IX). Although we have not characterized all products on the gel, based on size, we have indicated the likely origin of additional species.
- © 2012 Cold Spring Harbor Laboratory Press








