
A Versatile Protocol for mRNA Electroporation of Xenopus laevis Embryos
- Department of Biology and Center for Regenerative and Developmental Biology, Tufts University, Medford, Massachusetts 02155
Abstract
Xenopus laevis is an ideal model system for investigating the mechanisms of pattern formation. The ability to express exogenous mRNA or introduce morpholinos into cleavage-stage Xenopus embryos has allowed gain- and loss-of-function experiments that reveal molecular-genetic control of development and regeneration. However, injection of mRNAs into cleavage-stage embryos provides limited spatio-temporal control: It is difficult to limit targeting to small regions (e.g., inducing foci of expression) and the fate map does not facilitate targeting some tissues, such as those of the tail. Likewise, early injection can result in unwanted developmental defects because mRNA can be translated long before the desired time point. These are especially important limitations when studying developmental and regenerative processes during the gastrula to tailbud stages. Although transgenic techniques allow precise control over spatio-temporal expression of genes when the appropriate promoter is available, the process of creating stable transgenic animals is time-consuming. Electroporation provides an alternative method for delivering mRNA and other nucleic acids, enabling the targeting of single cells or groups of cells at any stage of development. This protocol describes detailed electroporation parameters for the transfection of mRNA into a wide range of tissues in embryos at gastrula to tailbud stages, with high efficiency and expression as early as 4 h post electroporation.
MATERIALS
Reagents
Agarose solution (1% prepared in 0.1× MMR)
Marc's modified Ringer's (MMR) (1×)
mRNA encoding protein of interest
-
For expression in Xenopus, capped mRNAs synthesized from CS2+ vectors are recommended. The vector is available from the authors upon request.
mRNA delivery solution (1–2 µg/μL mRNA in H2O and 0.2–3 mM Alexa Fluor 488 dextran 3000 or Alexa Fluor 555 dextran 3000 in H2O)
Tricaine methanesulfonate (MS222) (0.03% prepared in 0.1× MMR and adjusted to physiological pH)
Xenopus embryos/tadpoles at stage of interest
-
Xenopus embryo stages are designated according to Nieuwkoop and Faber (1967).
Equipment
Dissection microscope
Electroporation chamber (assembled as described in Steps 1–3)
Fluorescence microscope
-
A possible configuration would be Olympus BX-61 spinning disk confocal microscope, Hamamatsu ORCA AG CCD camera, MetaMorph software, and appropriate filter sets for visualizing fluorescent or fluorescently tagged proteins.
Forceps (fine; for breaking needles)
Injection needles (made from pulled capillary glass)
Injector (e.g., PLI-100 from Medical Systems)
Micromanipulator (for injection) (e.g., M3301R from World Precision Instruments)
Parafilm
Petri dishes (60 × 15 mm) (e.g., Fisherbrand)
-
A 60 × 15-mm Petri dish that has a precut 20-mm diameter center hole is used to prepare the electroporation chamber (see Step 1).
Pipettes (disposable transfer)
Pulse generator (e.g., CUY21EDIT [square wave electroporator] from Nepa Gene)
-
CUY21EDIT requires additional equipment including 20-mm diameter round platinum plate electrode (anode) in a 100 × 15-mm Petri dish (CUY700P20E), a needle cover platinum electrode (cathode) 0.3 mm, 20-mm tip (CUY195P0.3), and two cables connecting each electrode to the voltage plug terminal of CUY21EDIT. Be sure to read the safety information and instructions contained in the operating manual of the electroporation machine.
Silicone mold for making dimples in agarose
-
The dimples are arrayed, each 1.5 mm in diameter and 1 mm in depth, as shown in Figure 1. The original template for making silicone molds was cut from polystyrene plastic stock using a Roland MDX-40 three-axis computer numerically controlled milling machine using Roland proprietary Modela 4 software. Any two-part curing silicone rubber (e.g., MTT Technology Group's VTV 800 silicone molding compound) may be mixed and poured into the polystyrene template and allowed to cure to produce silicone molds.

A picture of the silicone mold used for making dimples in agarose. The dimples are 1.5 mm in diameter, 1 mm in depth, and arranged in an array. For a detailed explanation of how this mold is prepared, see the Equipment section.
METHOD
Assembly of Electroporation Chamber
This procedure takes ∼40 min.
-
1. Take a 60 × 15-mm Petri dish that has a precut 20-mm diameter center hole in the bottom and cover the outside with Parafilm.
-
2. Pour 1% agarose (prepared in 0.1× MMR) into the dish to a final thickness of 2–3 mm.
-
3. Immediately insert a silicone mold (Fig. 1) (for up to 20 pre-tailbud embryos) or 5-mm pieces cut from micropipette tips (for up to 7 tailbud/tadpole stage embryos) into the agarose bed to make pockets that will keep the animals in place during injection and electroporation (see Fig. 2). While the agarose cools and solidifies, which takes ∼30 min, proceed with Steps 4–7.
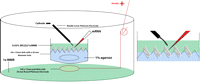
Schematic diagram showing set-up for the injection and electroporation apparatus. For detailed instructions, see Steps 1–7.
Set-up of Injection and Electroporation Apparatus
Ideally, the micromanipulator and electroporator should be set up on opposite sides (right and left) of the dissection microscope.
-
4. Adjust the micromanipulator so that the needle position is at an angle of ∼45° with respect to the base of the dissection microscope.
-
5. Calibrate the needle. For example, using fine forceps, break the needle so that it delivers 10–20 nL of solution per injection.
-
Use needles with a diameter of <1 µm at the tip to limit tissue damage and prevent mRNA leakage once the needle is removed following injection.
-
-
6. Fill the needle with the appropriate mRNA solution (e.g., 0.5 µL of 1–2 µg/μL concentration) using the “fill” button on the injector. Keep the needle tip in water to prevent clogging.
-
7. Set up the electroporation apparatus (see Fig. 2). When the agarose has solidified, remove the Parafilm from the bottom of the dish and the silicone mold or pipette tips from the agarose bed. Place the 60 × 15-mm dish in the round platinum electrode 100 × 15-mm Petri dish containing 30 mL of 1× MMR (or a large enough volume to cover the electrode).
Do not add the embryos at this step, but only after Step 10 is completed.
Determinination of Electroporation Parameters
The parameters that need to be determined are poring pulse (to form pores in the plasma membrane) and driving pulse (to drive mRNA into the cytosol) (Akaneya et al. 2005). This procedure takes ∼5 min.
-
8. Cover the agarose in the 60 × 15-mm injection/electroporation dish with 1× MMR. Place the cathode into the MMR and perform a resistance (ohm) measurement by pressing the “ohm check” button.
-
9. Depending on the resistance value measured in Step 8, set up the poring and driving pulse parameters according to Table 1.
-
In CUY21EDIT, poring and driving parameters can be stored in “Memory 1” and “Memory 2” and easily retrieved by simply pressing M1 and M2 buttons, respectively. See Troubleshooting.
-
Resistance-dependent poring and driving pulse parameters for mRNA electroporation
Injection and Electroporation
-
10. Anesthetize tailbud/tadpole-stage embryos by immersing them in 0.03% MS222 for ∼5 min.
-
Omit this step for embryos younger than the neurula stage.
-
-
11. Transfer the anesthetized animals into the injection/electroporation dish, carefully orienting each of them in the agarose bed pockets (see Fig. 2). Use 0.03% MS222 solution in the injection/electroporation dish to keep tailbud/tadpole-stage embryos anesthetized until completing Step 14.
-
12. Inject 10–20 nL of mRNA into the desired location and immediately proceed to Step 13.
-
The injection volume can be varied to target fewer or more cells. Injection/electroporation can be performed on early stage embryos without removing the vitelline membrane. The use of Alexa Fluor 488 dextran 3000 or Alexa Fluor 555 dextran 3000 will aid visualization of solution delivery and retention in tissue following injection. See Troubleshooting.
-
-
13. Position the cathode ∼1 mm away from the injection site (without the electrode touching the embryo) and apply poring and driving pulses, respectively.
-
14. Repeat Steps 12 and 13 until all the animals in the agarose bed pockets have been electroporated.
-
The injection and electroporation process takes ∼1 min per embryo or tadpole.
-
-
15. Transfer the animals to fresh 1× MMR and keep them in this solution for 1 h.
-
16. Transfer the animals to 0.1× MMR and keep them overnight at 14–18°C.
Scoring for Protein Expression and Imaging
-
17. Anesthetize tailbud/tadpole stage animals by immersing them in 0.03% MS222 for ∼5 min.
-
18. Mount anesthetized animals onto the stage of a microscope (to view and score developmental phenotypes) equipped with the necessary filter sets to visualize fluorescently tagged proteins encoded by the electroporated mRNA.
-
See Troubleshooting.
-
TROUBLESHOOTING
Problem (Step 9): The measured resistance values are out of range.
Solution: If resistance values are >18 kΩ, check to make sure that the electrodes are properly connected to the pulse generator. If resistance measurements range between 1 and 18 kΩ, consider lowering these values using an injection/electroporation medium with more concentrated MMR (i.e., higher ionic strength). Clean the platinum electrodes (both anode and cathode) regularly to avoid build-up on the electrodes that increases resistance. Follow the manufacturer's recommended cleaning and drying procedure for best results with the electrodes.
Problem (Step 12): Protein expression site does not match that of injection/electroporation site.
Solution: Electroporate immediately after injection because pressure-injected mRNA diffuses. Use a needle angled at ∼45° for accurate delivery of mRNA to target site.
Problem (Step 18): No protein expression is observed.
Solution: Increase the volume and/or concentration of the injected mRNA. To prevent RNase contamination and minimize mRNA degradation, use gloves while handling mRNA samples and keep mRNA on ice or in a freezer at −80°C.
DISCUSSION
This protocol provides poring parameters (high voltage, short duration, few pulses) that minimize macroscopic tissue damage and nonspecific embryonic defects. We present several sets of parameter values that take into account the resistance of the electroporating solution (see Table 1), which allow the procedure to be performed in concert with physiology or drug screening experiments in which the medium composition can be varied and also allow control over the size of the expressing region, facilitating studies of signaling by specific regions or cell groups.
Under our electroporating conditions, we achieve 86–93% transfection efficiency with 100% viability using stage 15–35 animals. In addition to the observed high rates of transfection and viability, our techniques allow for targeted expression in tissues such as the flank and tail, both of which are hard to get by cleavage stage injection (Fig. 3). The high voltage/short duration poring pulse and the low voltage/longer duration driving pulse parameters can also be used successfully for targeted endotoxin-free DNA and morpholino delivery (data not shown).
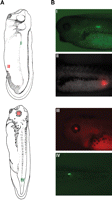
Generation of tdTomato- and EosFP-expressing Xenopus embryos via electroporation at stages 28 and 35. Illustrations in Panel A were retrieved from the Xenbase database (Bowes et al. 2009). (Reproduced with permission from Normal Table of Xenopus laevis (Daudin) by Nieuwkoop and Faber [1994]; ©1994 Garland Science/Taylor & Francis Books, LLC.) (A) Illustrations showing regions of EosFP (I and IV) and tdTomato (II and III) expression following electroporation. (B) Images showing expression in myotomes (I), hindgut (II), eye (III), and tail muscles (IV). These images were obtained with an Olympus BX-61 spinning disk confocal microscope equipped with a Hamamatsu ORCA AG CCD camera, controlled by MetaMorph software. Expression of EosFP (a green-to-red photoconvertible protein; Wacker et al. 2007) was imaged using the filters EX 470/20, BS 485, and EM 517/23 (Chroma filter set 41001). Expression of tdTomato (a red fluorescent protein; Shaner et al. 2004) was imaged using the filters EX 545/20, BS 565, and EM 595/50.
For further information regarding practical aspects of targeted delivery of mRNA, cDNA, and morpholinos to Xenopus embryos by electroporation, see Sasagawa et al. (2002), Falk et al. (2007), and Liu and Haas (2011).
ACKNOWLEDGMENTS
We thank Junji Morokuma and Kelly McLaughlin for helpful discussions, and Franz Oswald for the kind gift of EosFP construct. M.L. is grateful for support by the NIH (EY018168, AR061988, GM078484, AR055993), the G. Harold and Leila Y. Mathers Charitable Foundation, and the Telemedicine and Advanced Technology Research Center (TATRC) at the U.S. Army Medical Research and Materiel Command (USAMRMC) through award W81XWH-10-2-0058.
- © 2012 Cold Spring Harbor Laboratory Press








