
Imaging Neuronal Activity with Genetically Encoded Calcium Indicators
Adapted from Imaging in Neuroscience (ed. Helmchen and Konnerth). CSHL Press, Cold Spring Harbor, NY, USA, 2011.Abstract
Genetically encoded calcium indicators (GECIs), which are based on chimeric fluorescent proteins, can be used to monitor calcium transients in living cells and organisms. Because they are encoded by DNA, GECIs can be delivered to the intact brain noninvasively and targeted to defined populations of neurons and specific subcellular compartments for long-term, repeated measurements in vivo. GECIs have improved iteratively and are becoming useful for imaging neural activity in vivo. Here we summarize extrinsic and intrinsic factors that influence a GECI’s performance and provides guidelines for selecting the appropriate GECI for a given application. We also review recent progress in GECI design, optimization, and standardized testing protocols.
INTRODUCTION
Calcium ions are universal second messengers regulating essential cellular signaling events in a wide range of tissues and organisms. In neurons, action potentials (APs) trigger large and rapid calcium influx through voltage-gated channels. Similarly, activation of neurotransmitter receptors during synaptic transmission causes calcium transients in dendritic spines. With appropriate indicators, imaging intracellular calcium dynamics can be used to measure neuronal spiking and synaptic activity across populations of neurons in vitro and in vivo.
There are many fluorescence imaging techniques for visualizing calcium dynamics in living cells and animals. Bulk or intracellular loading of synthetic calcium indicators has been used widely (Yuste et al. 1992; Svoboda et al. 1997; Fetcho et al. 1998; Stosiek et al. 2003). However, small-molecule indicators have many drawbacks that prevent their applicability for a wide variety of questions in neuroscience. They cannot easily be targeted to specific cell types, populations, or subcellular locations (but see Tour et al. 2007). Because loading is highly invasive and can be damaging to tissue, they also are incompatible with repeated, chronic in vivo measurements.
GECIs based on chimeric fluorescent proteins address many of these limitations and provide an alternative to small-molecule calcium dyes. GECIs can be targeted to specific cell types, populations, or subcellular compartments, allowing the imaging of structures such as synapses, axons, and dendrites (for review, see Palmer and Tsien 2006). The delivery of GECIs into intact tissues and organisms (e.g., by using transgenesis or viral constructs) is minimally invasive and therefore compatible with long-term, repeated in vivo measurements (Mank et al. 2008), although calcium buffering and cellular perturbation can occur.
In general, GECIs consist of a calcium-binding domain (e.g., calmodulin or troponin C), fused to one or two fluorescent proteins (FPs) (Fig. 1A–C). In single-FP GECIs, the fluorescence intensity of a circularly permuted or split FP is modulated by calcium binding–dependent changes in the chromophore environment (Baird et al. 1999; Nagai et al. 2001; Nakai et al. 2001). In two-FP GECIs, calcium binding allosterically modulates the relative donor–acceptor emission spectra through a distance- and orientation-dependent change in fluorescence resonance energy transfer (FRET) (Miyawaki et al. 1997; Heim and Griesbeck 2004; Palmer et al. 2006). In many cases, a conformational actuator (e.g., the M13 peptide) is included in the fusion protein to enhance the conformational change and fluorescence modulation.
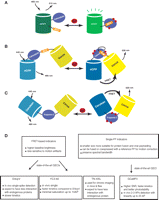
Schematic representation of the three major GECI classes. GECIs are based either on florescence intensity changes of split or circularly permuted single FPs (A) or changes in FRET efficiency between two FPs (B,C). (A) Schematic of the G-CaMP-type sensing mechanism. On calcium binding, conformational changes in the calmodulin–M13 complex induce fluorescence changes in the circularly permuted enhanced GFP (cpEGFP). (B) The cameleon family of FRET-based GECIs. A calcium-dependent increase in FRET between a CFP and YFP FRET pair is coupled to the binding of calmodulin to the M13 peptide. (C) Troponin C–based FRET GECIs. Binding of calcium to troponin C induces conformational changes and an increase in FRET between CFP and YFP. (D) Practical considerations for choosing the most appropriate GECI.
Through iterative cycles of optimization, GECIs have been improved to the point that they are useful for in vivo neuronal imaging.
-
The calmodulin-based FRET sensor D3cpVenus (D3cpV; an improved cameleon variant) (Palmer et al. 2006) has been used to detect single APs in organotypic mouse brain slices and in vivo in layer 2/3 somatosensory cortical neurons (Wallace et al. 2008).
-
Recently, another cameleon variant, YC3.60 (Nagai et al. 2004), allowed detection in vivo of single APs with a sensitivity comparable to that of D3cpV, but with faster kinetics and minimal saturation up to 10 APs (Lütcke et al. 2010).
-
The troponin C–based FRET sensor TN-XXL has been used for chronic in vivo activity imaging in mouse and fly (Mank et al. 2008).
-
The single-FP sensor G-CaMP3 showed high expression and sensitivity with no apparent cytotoxicity or behavioral phenotype in Caenorhabditis elegans and Drosophila (Tian et al. 2009). G-CaMP3 allowed the detection of single APs in pyramidal neurons in acute mouse brain slices. The long-term imaging (months) with G-CaMP3 in the motor cortex of behaving mice showed large fluorescence changes (detecting two to three APs) with linearity up to 20 APs (Tian et al. 2009).
Characterizing several GECIs in the context of a specific application is important, because they have different strengths and limitations (Fig. 1D). To help guide GECI selection and experimental design, we have analyzed factors affecting GECI performance, and we review recent progress in GECI optimization.
PROPERTIES THAT INFLUENCE THE PERFORMANCE OF GECIs
A uniform standard for comparing the performance of calcium indicators is the signal-to-noise ratio (SNR). The SNR is defined as the ratio of the fluorescence signal change (ΔF = Fobs − F0, where Fobs is sensor fluorescence at peak [Ca2+] and F0 is sensor fluorescence at baseline [Ca2+]) to the shot noise on the baseline fluorescence (F0N−1/2, where N is the number of photons detected) (Yasuda et al. 2004). An equivalent calculation for FRET sensors takes the difference in donor–acceptor ratio divided by the combined shot noise on the baseline fluorescence of both FPs.
To optimize SNR, the appropriate combination of intrinsic GECI parameters should be matched with the extrinsic factors of the system studied. Extrinsic factors include the size, time course and frequency of calcium transients, and basal [Ca2+] of the target cell type in the organism studied. Intrinsic GECI parameters include the sensor affinity, kinetics, dynamic range, brightness, expression level, fluorescence properties, and independence from endogenous interference.
Underlying Calcium Dynamics
Neurons typically maintain extremely low cytoplasmic-free [Ca2+] (30–200 nm) at rest (Hille 1992). When stimulated, calcium flows into the cytoplasm through calcium channels (voltage-gated calcium channels or calcium-permeable, receptor-operated channels) or is released from calcium-loaded organelles, rapidly raising the local [Ca2+] (Hille 1992). Calcium levels then return to baseline via extrusion from the cell and reloading of intracellular buffers and stores. The spatiotemporal evolution of calcium transients is shaped by the localization, mobility, affinity, and kinetics of these processes, including buffering by endogenous proteins and any exogenous GECI present (Helmchen et al. 1996).
In neurons, APs occur over a wide range of frequencies. When sparse, each induced calcium transient is independent, fast (<2 msec rise time), and modest (∼250 nm in pyramidal neurons) (Helmchen et al. 1996). When closely spaced, the calcium transients accumulate (Helmchen et al. 1996). Depending on the application, users can choose to optimize any of the following: detection of sparse APs, dynamic range of burst detection, or temporal resolution of individual APs in spike trains. This guides the GECI selection as discussed below; for a detailed treatment, see Hires et al. (2008a).
Calcium Affinity, Kinetics, and Dynamic Range
The fluorescence response of a GECI to calcium transients is critically dependent on the affinity, kinetics, and dynamic range of the sensor. Sensors with a linear relationship between fluorescence response and target stimulus are preferred for quantitative measurement of calcium transients (Yasuda et al. 2004). Nonlinearity in sensor or cellular calcium dynamics can prevent precise spike quantification (e.g., firing rate and the number of APs) during action potential bursts (Sasaki et al. 2008).
To obtain large signal changes in response to small calcium transients (such as those triggered by sparse AP firing), a high-affinity sensor is preferred. However, such a sensor will tend to buffer more Ca2+ and can influence the kinetics and amplitude of transients during chronic expression (Mank and Griesbeck 2008). It will also saturate more easily, decreasing the linearity at high calcium concentrations, such as those resulting from long or closely spaced spike trains. A combination of high affinity and fast kinetics is optimal for a sensor to resolve incremental signal increases with additional APs. However, it is challenging to maintain both in GECI engineering because affinity is inversely dependent on the dissociation rate (Mank et al. 2006). A low affinity provides a baseline F0 (fluorescence at resting [Ca2+]) closer to Fmin (fluorescence at zero [Ca2+]), and therefore a greater effective dynamic range (the maximum signal change at saturating [Ca2+]). Thus, such a sensor is useful for the quantitative measurement of larger calcium transients (e.g., those through synaptic neurotransmitter receptors or from high-frequency spike trains).
It is important to calibrate and compare the calcium affinity and dynamic range of each GECI under the same experimental conditions because those parameters can be influenced by external factors such as total ionic strength (Linse et al. 1991), pH, temperature, and presence of competing ions (e.g., Mg2+) (Ogawa and Tanokura 1984; Hires et al. 2008a).
GECI Expression Level
GECIs, and other kinds of calcium monitors, bind calcium and thus inherently add to calcium buffering. Higher [GECI] improves the SNR by increasing the number of photons detected. However, very highly expressed GECIs can become dominant calcium buffers and spread transients over longer times and lower amplitudes, thus changing the calcium signaling in cells (Helmchen et al. 1996). Early work on Yellow Cameleon (YC) showed that HeLa cells expressing YC3.1 at <20 µm had poor SNR, whereas cells with [YC3.1] >300 µm showed significant calcium buffering (Miyawaki et al. 1999). Similarly, in G-CaMP2 imaging in acute mouse cortical slices, cells with [G-CaMP2] <5 µm required unacceptably high laser intensity, whereas cells with [G-CaMP2] >20 µm showed attenuated responses for short AP trains (Hires et al. 2008a).
To estimate [GECI], the fluorescence of the purified sensor can be compared with that of the expressed sensor under identical imaging conditions (Tour et al. 2007). For FRET-based sensors, the emission of the directly excited acceptor chromophore is used because this is theoretically insensitive to calcium levels (Miyawaki and Tsien 2000). For intensity-modulated single-FP sensors, [GECI] estimates can vary with resting calcium levels in different systems. Estimates can be made by calibration with a second calcium dye (Tay et al. 2007), application of ionomycin or calcium chelators (McCombs and Palmer 2008), or knowledge of the basal spike rates and calcium levels. With all methods, misfolded, constitutively fluorescent, or nonfluorescent GECI forms can contribute to errors in the estimates of [GECI] and calcium buffering.
In addition to buffering Ca2+, GECI components (e.g., calmodulin, M13) can be sequestered by endogenous proteins, resulting in high background and nonfunctional indicators (Hasan et al. 2004; Heim and Griesbeck 2004). GECIs constructed with components mutated relative to endogenous calcium-signaling molecules (Palmer et al. 2006) or with calcium-binding proteins not typically found in target cells (Heim and Griesbeck 2004) can reduce such interference.
Both altered calcium handling and endogenous interference can perturb cell-signaling pathways and lead to behavioral phenotypes. For example, C. elegans expressing G-CaMP3 in chemosensory neurons showed local search turning behavior similar to that of wild-type animals. However, decreased turning was observed in animals expressing YC3.60, YC2.12, G-CaMP1, and G-CaMP2 (Tian et al. 2009).
GECI Fluorescence Properties
Intrinsic factors influencing the GECI performance include brightness (extinction coefficient × quantum yield), pH sensitivity, folding stability, and photobleaching of the sensor.
Substituting FPs with enhanced fluorescence properties has proven to be a reliable method of improving sensor brightness and signal change. In the cameleon family, when yellow fluorescent protein (YFP) was substituted by Venus (Nagai et al. 2002) or Citrine (Griesbeck et al. 2001), the resulting sensors showed lower pH sensitivity and improved folding efficiency. The maximum FRET change was improved further to ∼500% in vitro by using a circularly permuted Venus as the FRET acceptor (Nagai et al. 2004). In the troponin family, the sensor’s brightness and FRET change have been optimized by substitution of both cyan fluorescent protein (CFP) with its brighter variant Cerulean (Heim et al. 2007) and YFP with circularly permuted Citrine (Mank et al. 2006). In the G-CaMP family, incorporating a subset of the “superfolder green fluorescent protein (GFP)” mutations (Pédelacq et al. 2006) improved brightness at physiological temperatures (Tallini et al. 2006).
The state-of-the-art GECIs include G-CaMP3, D3cpV, YC3.60, and TN-XXL. In vitro dynamic ranges are 560%, 400%, and 200% FRET increases for D3cpV, YC3.60, and TN-XXL (Mank and Griesbeck 2008), respectively, and a 1200% fluorescence intensity increase for G-CaMP3 (Tian et al. 2009). In neurons, the smaller fluorescence changes produced by the FRET indicators resulted in lower SNR than G-CaMP3 (Tian et al. 2009), although they are brighter at submicromolar [Ca2+].
Improved photostability increases the duration that activity can be continuously monitored. G-CaMP3 showed greater photostability than the FRET sensors D3cpV and TN-XXL (Tian et al. 2009), presumably because the dark basal state is less susceptible to bleaching. For both intensity and FRET imaging, GECIs whose signals increase on Ca2+ binding are preferable, because this reduces misinterpretation of potential artifacts such as sensor bleaching, degradation, or hardware errors. The comparison of GECI performance in acute slice is shown in Table 1.
Properties of the state-of-the-art GECIs in acute mouse brain slices
GECI OPTIMIZATION
Practical Improvement
Protein engineering efforts have significantly improved the properties of several genetically encoded indicator classes. Overall, sensor brightness and fluorescence changes have been improved by substituting superior FPs (Griesbeck et al. 2001), linker truncation (Deuschle et al. 2005; Hires et al. 2008b), and FP circular permutation (Nagai et al. 2004) and by site-directed and random mutagenesis (Tallini et al. 2006). The computational protein design of “bump-hole” calmodulin/M13 pairs (Palmer et al. 2006) can decrease interference with endogenous proteins (Palmer et al. 2006). Alternatively, troponin C, a skeletal calcium sensor not normally expressed in neurons, has been used in place of calmodulin (Heim and Griesbeck 2004). Affinity and specificity of binding domains for Ca2+ have also been tuned by the rational design (Mank et al. 2006, 2008).
From G-CaMP2 to G-CaMP3
We recently reported the improvement of the single-FP GECI G-CaMP2 (Nakai et al. 2001; Tallini et al. 2006). Our aims for maximizing the SNR of the G-CaMP scaffold were threefold: increased fluorescence change to small and rapid calcium transients, larger dynamic range, and increased baseline brightness.
To optimize G-CaMP, we used semirational library screening guided by the G-CaMP2 protein structure (Wang et al. 2008; Akerboom et al. 2009). To increase baseline brightness, we targeted positions that improve protein folding and stability. To increase the dynamic range, we mutagenized sites near the enhanced GFP chromophore. To increase the calcium affinity and allow for better detection of sparse APs, we mutated the Ca2+-binding EF hands and the interface between the M13 peptide and calmodulin.
Relative to G-CaMP2, the improved variant, named G-CaMP3, is three times brighter, possesses greater protein stability, and has three times the dynamic range and a higher affinity (1.3×) for calcium (Fig. 2A; Tian et al. 2009). G-CaMP3 detected single-AP–triggered calcium transients in pyramidal cell dendrites, with SNR, response linearity, and photostability significantly better than those of G-CaMP2 (Fig. 2B). In C. elegans chemosensory neurons, and the Drosophila antennal lobe, sensory stimulation-evoked fluorescence responses were significantly enhanced with the new indicator (4×–6×), and no apparent cytotoxicity was detected (Fig. 2B). In somatosensory and motor cortical neurons in the awake, behaving mouse brain, the sensor detected three to 10 AP trains with a good response linearity and photostability. It also allowed long-term repeated imaging of targeted neuronal populations in the motor cortex of behaving mice (Fig. 2C; Tian et al. 2009). G-CaMP3 plasmid DNA in a eukaryotic expression vector is now available from AddGene. AAV-G-CaMP3 constructs and live virus are available from the University of Pennsylvania Vector Core (http://www.med.upenn.edu/gtp/vectorcore/); fly and worm stocks are available on request.
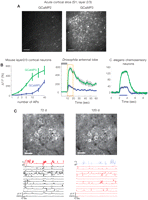
Imaging neural activity with improved G-CaMPs (Tian et al. 2009). (A) The improved baseline fluorescence of G-CaMP3 compared with that of G-CaMP2. Both indicators were virally delivered to layer 2/3 cortical neurons, and images were taken 12 d post-viral injection. Scale bar, 50 µm. (B) The average fluorescence change of G-CaMP3 is greater than that of G-CaMP2 in response to trains of APs given at 83 Hz in layer 2/3 cortical neurons (left). Sensory stimulation-evoked fluorescence responses of G-CaMP3 were significantly enhanced in the Drosophila antennal lobe (middle) and in C. elegans chemosensory neurons (right). (C) G-CaMP3 is suitable for long-term imaging of behaviorally correlated activity in neuronal populations over extended periods of time. Repeated imaging of the same neuronal population (L2/3 neurons of the primary motor cortex) at 72 (left) and 120 d (right) postinfection showed remarkably constant (top) G-CaMP3 expression (scale bar, 30 µm) and (bottom) signal change (DF/F of individual cells; black lines). (Red line) Relative treadmill movement (F, forward; B, backward). Scale bars, 20 µm. (Adapted, with permission, from Tian et al. 2009.)
GECI Expression
A GECI’s expression level influences its performance in cells and organisms. Low GECI expression produces poor SNR and difficulty in visualizing positive cells. High expression increases baseline brightness and SNR, but can alter calcium homeostasis and reduce the signal change. Prenatal expression (induced by in utero electroporation of G-CaMP3, D3cpV, or TN-XXL driven by the strong CAG promoter) altered subcellular sensor distribution and neuronal excitability in some instances (Tian et al. 2009). Postnatal expression of G-CaMP3 from the weaker synapsin-1 promoter via viral-mediated gene transfer alleviated many of these effects (Tian et al. 2009). For each application, GECI expression timing and magnitude should be optimized to balance signal and cytotoxicity by testing multiple promoters, regulatory sequences, and transduction methods (e.g., screening different viral serotypes).
Subcellular Targeting
GECIs can be targeted to specific subcellular locations by protein fusions or signaling peptides. This could reveal underlying Ca2+ dynamics in particular organelles (e.g., the pre- and postsynapse) and might also improve the SNR of the AP detection without altering the sensor itself. For example, a low-affinity GECI might be less useful for probing small calcium transients in the cytosol but highly effective at measuring large calcium transients in the endoplasmic reticulum (Miyawaki et al. 1999). For neurons showing small somatic calcium changes during spiking, a high-affinity GECI is required. Alternatively, GECIs can be targeted to presynaptic terminals, where Ca2+ flux is high, to increase the signal change associated with APs (Dreosti et al. 2009). The increase of Ca2+ could be even larger in the vicinity of calcium-channel microdomains or synaptic vesicles (Roberts 1994). GECIs can also be targeted to the nucleus (Miyawaki et al. 1997), which might provide an efficient solution for facile segmentation in image processing (L. Looger, unpubl.).
GECI TESTING STANDARDIZATION
The performance of available GECIs has been compared in a wide variety of systems, both in vitro (including purified protein, cultured cells, and hippocampal neurons [Palmer et al. 2006; Tian et al. 2009], and mouse brain slice [Pologruto et al. 2004; Mao et al. 2008; Tian et al. 2009]), and in living animals including worm chemosensory neurons (Tian et al. 2009), Drosophila antennal lobe (Tian et al. 2009) and larval neuromuscular junction (Reiff et al. 2005; Hendel et al. 2008), and rodent cortical layer 2/3 pyramidal cells (Mank et al. 2008; Wallace et al. 2008; Tian et al. 2009; Lütcke et al. 2010). Because each system or cell type has a different calcium signaling tool kit, a GECI with high sensitivity in one setting might not be the best fit when conditions change. The GECI performance in vitro correlates poorly with that in more intact preparations (Tian et al. 2009). Therefore, we sought to establish a standardized, multistep screening paradigm for GECI developers.
For developmental efficiency, the initial testing system should allow screening for brightness, calcium sensitivity, dynamic range, and kinetics with high throughput. It should also capture as many aspects of intact preparations (e.g., calcium buffering and cytopathicity) as possible. Our method uses agonized endogenous G-protein coupled receptors in cultured mammalian cells to create synthetic Ca2+ transients lasting tens of seconds (Tian et al. 2009). Incorporating neurotransmitter receptors into mammalian cells mimics the fast calcium transient in neurons, providing better kinetic sensitivity (L. Looger, unpubl.). The use of alternative cell types could obscure the dynamic aspects of Ca2+ signaling unique to neuronal morphology and physiology; emphasis must be placed on systematic analyses in neurons and eventually in intact animals. The recent development of microfluidic cell sorter devices could facilitate library screens of GECI with single-cell resolution (A. Palmer, pers. comm.), although this could restrict the choice of cell types and depolarizing stimuli.
The two-photon imaging of GECI responses to back-propagating APs in acute brain slices captures many aspects of in vivo functional imaging (e.g., possible long-term expression and developmental effects) while allowing a moderate throughput. Therefore, it establishes a good standard for comparing a small number of high-quality sensors after an initial screen. Defined numbers of APs can be triggered at specific intervals by intermittent short depolarizing pulses (Pologruto et al. 2004). Alternatively, neurons can be depolarized by continuous current injection (Hendel et al. 2008) from a patch pipette. The calcium transients associated with back-propagating APs have been well characterized. Because the GECI is introduced prenatally by in utero electroporation, potential interference with development should be captured (Mao et al. 2008; Tian et al. 2009).
Owing to its greater imaging depth, motion artifacts, and hemodynamics, in vivo imaging has more challenging SNR requirements. Therefore, it is necessary to confirm the performance of the best candidates in in vivo preparations. Also, to better help end-users choose the most appropriate GECI, the in vivo performance of state-of-the-art GECIs should be compared under identical conditions (e.g., expression cassette, expression time and level, cell type, stimuli).
USING GECIs TO MEASURE NEURAL ACTIVITY
Quantitative neural activity measurements in vivo require calibration of the GECI response to electrophysiological measurement of spike trains in an example set of neurons. In the mammalian brain, calibration via cell-attached recording of spontaneous APs is preferred. Further efforts (e.g., whole-cell patch clamp or synaptic input mapping in slice) should be made to verify that GECI expression does not significantly alter the neurons’ physiological properties or network interactions (Tian et al. 2009).
Analytical techniques developed for quantitative imaging with small-molecule calcium dyes can be adapted to GECI imaging, provided the response parameters of the GECI (ΔF/AP, linearity) are determined. Possibilities include reconstruction of firing rate changes via deconvolution (Yaksi and Friedrich 2006), machine-learning algorithms (Sasaki et al. 2008), or Monte Carlo spike detection (Vogelstein et al. 2009).
THE FUTURE OF GECI DESIGN
The primary focus of current GECI engineering efforts is to further increase the SNR of existing GECIs for the reliable detection of low-firing rates in neurons. Engineering high-affinity sensors for probing small stimuli while preserving fast kinetics is challenging. To balance high SNR with minimal cytotoxicity, expression cassettes that are inducible and reversible or that hold [GECI] steady for long periods of time would facilitate signal calibration and further reduce toxicity concerns in chronic imaging. Careful experiments are required to determine the effect of long-term GECI expression on cells and circuits. Finally, novel GECI scaffolds could be engineered for even greater performance and broader application in neural activity imaging.
ACKNOWLEDGMENTS
We thank Daniel Huber, Jasper Akerboom, Vivek Jayaraman, and Karel Svoboda (all at Howard Hughes Medical Institute [HHMI] Janelia Farm) for valuable discussions.
- © 2012 Cold Spring Harbor Laboratory Press








