
Using the RCAS-TVA System to Model Human Cancer in Mice
- Program in Gene Function and Expression, University of Massachusetts Medical School, Worcester, Massachusetts 01605
Abstract
For successful infection, avian sarcoma leukosis virus subgroup A (ASLV-A) requires its receptor, tumor virus A (TVA), to be present on the surface of target cells. This is the basis of the RCAS-TVA gene delivery system: Mammalian cells lack the gene encoding TVA and are normally resistant to infection by ASLV; however, transgenic targeting of TVA to specific cell types or tissues in the mouse renders these cells uniquely susceptible to infection by ASLV-A-based RCAS viruses. The RCAS-TVA system is a powerful tool for effectively modeling human tumors, including pancreatic, ovarian, and breast cancers, gliomas, and melanomas. RCAS viruses can deliver cDNAs (≤2.8 kb), as well as short hairpin RNAs (shRNAs), microRNAs (miRNAs), and other noncoding RNAs. Compared with traditional transgenic and knockout mice, the RCAS-TVA system has several strengths. First, virus delivery is generally performed postnatally and results in a relatively low infection rate of target cells; the sporadic postnatal expression of the gene of interest mimics the situation in developing human tumors. Second, a single transgenic mouse line can be used to compare the consequences of specific genes on tumor development, with viruses encoding oncogenes or shRNAs targeting specific tumor suppressor genes. TVA mouse strains can also be easily combined with transgenic, knock-in, and knockout mouse models to study cooperating genetic events.
The RCAS Virus
RCAS stands for replication-competent avian sarcoma-leukosis virus (ASLV) long-terminal repeat (LTR) with splice acceptor. The virus enters cells via the TVA receptor, which mammalian cells do not natively express. Following entry into a mammalian cell, a cDNA copy of the viral genome is made. When the cell undergoes division, the viral cDNA copy incorporates into the mammalian host genome.
The RCAS virus retains all of the necessary viral genes for replication. Therefore, in an avian cell culture, there is no need for the use of helper plasmids to generate virus as is commonly done with retroviruses that have tropism for mammalian cells. The immortalized chicken fibroblast cell line DF1 is commonly used to propagate RCAS viruses in culture (Himly et al. 1998; Schaefer-Klein et al. 1998). Chicken embryo fibroblasts may also be used. In addition, high-titer virus can be produced for several days or weeks following a single transfection of a viral plasmid into avian producer cells. (See Protocol: Generation of High-Titer RCAS Virus from DF1 Chicken Fibroblasts [Ahronian and Lewis 2014].) The virus glycoprotein binds to its receptor to gain entry into a cell. Because only avian cells naturally express the TVA receptor to the RCAS glycoprotein, in the absence of engineered TVA expression infection of mammalian cells is very limited.
Inserts in the RCAS retrovirus are stable up to 2.8 kb but less stable when >2.8 kb. It is common beyond this insert size that RCAS will lose its insert, generate truncation mutants, or produce low-titer virus. The limited carrying capacity of RCAS viruses is one of the major drawbacks of the RCAS-TVA system.
In tumor modeling, RCAS viruses have most commonly been used to deliver cDNAs encoding oncoproteins (Fig. 1). However, RCAS viruses can readily deliver dominant-negative versions of tumor-suppressor proteins (Du et al. 2007), shRNAs (Bromberg-White et al. 2004; Seidler et al. 2008), miRNAs, and other noncoding RNAs. Work by Pao et al. showed that the RCAS system could be adapted for use with tetracycline-regulated gene-expression systems, and Holmen and Williams showed the utility of this system in vivo in a KRAS-driven glioma model (Pao et al. 2003; Holmen and Williams 2005).
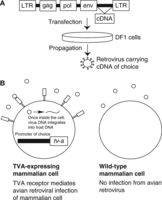
The RCAS-TVA system for mammalian cell infection. (A) Schematic of the RCAS retrovirus construct. This construct is transfected into DF1 chicken fibroblasts, where it is replication-competent and is produced at high titers. (B) RCAS viruses enter mammalian cells engineered to express the RCAS receptor, TVA. The reverse-transcribed retroviral genome integrates into the genome of the infected cell. TVA-negative mammalian cells are not infected by the RCAS virus.
Expression of the cDNA is typically mediated by viral LTRs (Fisher et al. 1999), but RCAS variants have been generated that allow expression to be driven by tissue-specific promoters or robust ubiquitous promoters. Modifications have been made to the RCAS plasmid to further control expression levels from the viral construct. RCAN is a modified version of RCAS where the splice acceptor has been removed (Hughes and Kosik 1984). Because the splice acceptor is required for expression from the viral LTR, a promoter of choice must be cloned into the RCAN virus in addition to the desired cDNA to attain expression. RCAN allows for customization of expression, which adds additional levels of control and context (Petropoulos and Hughes 1991).
The RCOS virus was developed to modify the level of expression of the inserted cDNA. Instead of the RCAS LTR sequence mediating expression of the inserted cDNA, expression is driven by the LTR from the RAV-0 virus. The RAV-0 LTR is a weaker promoter and yields lower levels of expression, ∼1/30–1/50 of the typical RCAS LTR level (Greenhouse et al. 1988).
To attain higher levels of RCAS viral titer, the polymerase region from RCAS was replaced with the sequence from Bryan RSV. This increases both the titer of RCAS viruses and the expression levels from the RCAS viral LTR. RCAS or RCAN plasmids that contain the Bryan polymerase are called RCASBP and RCANBP (Petropoulos and Hughes 1991). Given the improved titer and expression levels, this form of RCAS virus is most commonly used for in vivo experimentation.
Difficulties experienced with RCASBP/RCANBP are mostly related to cloning the appropriate cDNA insert due to the restricted number of unique restriction enzyme sites in the multiple cloning site of the plasmid. To facilitate cloning RCAS plasmids, a modified plasmid, RCAS-Y, containing a multiple cloning site linker was generated. Additionally, adapter plasmids were generated, which contain common restriction enzyme sites flanked by the restriction sites found in the RCASBP and RCAS-Y plasmids. Some of these adapter plasmids also include ATG sequences or splice acceptors to allow for further customization (Hughes et al. 1987). More recently, gateway cloning–compatible RCAS-Y (Loftus et al. 2001) and RCANBP (Bromberg-White et al. 2004) plasmids have been generated.
Because cell division is required for incorporation of the viral cDNA into the host genome, infection rates in vivo for most cell types and tissues are highest at the neonatal stage. Infection efficiency is greatly reduced after this period (Fisher et al. 1999). The cycling characteristics of the targeted cell type dictate how and when the RCAS virus is delivered in the mouse (see Protocol: In Vivo Delivery of RCAS Virus to Mice [Ahronian and Lewis 2014]). Difficulties with targeting cells that are nonproliferating at the desired time of infection can be circumvented with the use of lentiviral vectors pseudotyped with the ASLV-A envelope protein. This pseudotyped lentivirus efficiently infects nondividing or cell cycle–arrested cells, yet retains its dependence on TVA expression for infection (Lewis et al. 2001). This lentivirus was also used to induce breast cancer in vivo (Siwko et al. 2008).
In summary, there are multiple derivations of the RCAS viral vector that modify spatiotemporal expression, expression level, viral titer, and the ability to infect nondividing cells. Therefore, despite an insert size limitation of 2.8 kb, the viruses generated using RCAS are customizable for virtually any mouse modeling need.
Tumor Virus A
The tumor virus A (TVA) receptor was originally cloned from both chicken and quail DNA. TVA has two alternatively spliced isoforms, generating either a GPI-linked receptor or a transmembrane receptor. Expression of either isoform in mammalian cells is sufficient to permit infection by ASLV-A and RCAS viruses (Bates et al. 1993). Without the presence of the TVA receptor, infection of mammalian cells by RCAS viruses is extremely rare (<1 × 10−6) (Fisher et al. 1999). Expression levels of TVA in vivo can vary and still result in infection. It has been shown that low or moderate TVA levels may be superior to higher expression (Federspiel et al. 1994). It has been postulated that this effect results from the shedding of the GPI-linked receptor and its subsequent action as a decoy receptor.
Ubiquitous promoters, as well as tissue-specific and lineage-specific promoters, have been used to drive TVA expression in vivo and yielded successful RCAS infection. Table 1 shows a list of published transgenic TVA mouse strains, their tissue target, and tumors that have been modeled with that strain.
Published RCAS-TVA animal models
Advantages and Shortcomings of the RCAS-TVA System
There are several advantages of RCAS-TVA mouse models. RCAS viruses are replication competent in avian cells, but are replication defective in mammalian cells. Thus, there is no cell-to-cell spread after initial infection. The precise mechanisms that prevent virus production in mammalian cells are unknown. However, mRNAs encoding the viral proteins gag, pol, and env are undetectable in infected mammalian cells, and only the spliced message encoding the inserted gene of interest is detectable. Combined with the relatively inefficient infection of TVA-expressing mammalian cells in vivo, this results in a minority of the target cells being infected. This models the clonal origin of human solid tumors and preserves critical signaling between a mutant cell and wild-type neighboring cells that is important in human tumor-development, but is lost in other mouse model systems in which there is oncogene expression or tumor-suppressor inactivation throughout a target tissue. Because the viral genome integrates into the host genome, clonal analysis of resulting tumors can also be readily performed to determine their monoclonal or polyclonal nature. The replication deficiency of RCAS viruses in mammalian cells additionally limits a potential immune response that may occur with viral production in vivo.
Another benefit of RCAS-TVA models is that the effects of multiple genes on tumorigenesis can be assessed using a single TVA mouse strain instead of generating a mouse line for each gene of interest. This reduces the number of mouse strains that need to be generated and maintained, reducing costs and preserving animal colony space, which may be limited. For example, using a single mouse strain in which the elastase-promoter-directed TVA expression, three distinct pancreatic tumor types, reflecting the three main cell lineages in the pancreas, were induced depending on whether RCAS-c-myc or RCAS-PyMT was delivered (Lewis et al. 2003).
A key feature of the RCAS-TVA system is that it provides combined spatial and temporal regulation of gene expression by targeting specific cell types via TVA and temporal regulation via timed delivery of RCAS viruses. Previously, exploitation of the temporal feature of the system was restricted by the need for target cells to undergo cell division in order for the viral genome to integrate into the host genome. This commonly required RCAS virus delivery to neonatal mice to achieve sufficient infection of the target tissue. However, an ALSV-A-pseudotyped lentivirus was generated, which allows for infection in the absence of cell division (Lewis et al. 2001). Cell types and tissues that were previously difficult to target may now be able to be infected using the lentiviral system.
In addition to their utility in studying tumor-initiating lesions, RCAS-TVA models may also be used as efficient systems for the evaluation of potential cooperating genetic alterations. With the completion of several cancer genome sequencing projects, such studies will be of great value in ascertaining the pathogenic nature of identified gene mutations. For example, the desired TVA strain can be crossed to a mouse strain that initiates tumorigenesis, but that fails to rapidly progress to advanced disease. RCAS viruses encoding the candidate gene of interest can be delivered and assessed for their ability to promote tumor progression. Seidler et al. showed proof of principle by showing that RCAS-mediated delivery of p53-targeting shRNAs to mice with pancreas-specific expression of a KrasG12D allele promoted the development of PDAC (Seidler et al. 2008). Potentially, libraries of cDNAs and/or shRNAs in RCAS viruses could be used in large unbiased screens to identify cooperating genes in vivo as suggested previously for the study of breast carcinogenesis (Du and Li 2007).
In addition to in vivo tumor studies, the RCAS-TVA system can be used to infect primary cells in culture (Fisher et al. 1999). Cells can be isolated from a mouse expressing TVA in the desired population. These cells can be used for various assays in vitro or be transplanted into the mouse for tumor studies. This approach has been used for mammary epithelial cells (Park et al. 2001), ovarian surface epithelial cells (Orsulic et al. 2002), and pancreatic duct epithelial cells (Morton et al. 2007). These in vitro experiments can be used in conjunction with an RCAS-TVA cancer mouse model to provide mechanistic data in support of observed in vivo phenotypes.
The major weaknesses of the RCAS-TVA system surround the limited insert capacity of the virus and the relatively inefficient infection observed in vivo. The restriction of insert size to 2.8 kb in RCAS (less in RCAN vectors) limits the genes that can be evaluated with this system and excludes key oncogenic factors involved in human cancer. For example, the cDNAs for EGFR and Neu exceed the carrying capacity of RCAS vectors. Thus, prior studies evaluating these oncogenes have used truncated and activated versions of these oncogenes. Importantly, the limited carrying capacity of RCAS prevents this system from being used to evaluate tumor-associated mutations in EGFR that have been identified in NSCLC patients and which confer sensitivity to EGFR inhibitors currently in clinical use. The production of ASLV-A pseudotyped lentiviruses reduces this weakness somewhat, but the low viral titers produced with this lentiviral system limit its in vivo applications.
The other significant weakness of the RCAS-TVA system reflects the limited number of cells of the targeted tissue or cell type that become infected in vivo. Seidler et al. showed that intraperitoneal injection of 50 million DF1 producer cells to transgenic mice with pancreas-restricted expression of TVA results in the infection of <1% of pancreatic cells (Seidler et al. 2008). This low infection rate means that only very potent oncogenes are likely to stimulate tumor formation in vivo. Indeed, although delivery of DF1 cells producing RCAS-PyMT induced pancreatic cancer precursor lesions in vivo, delivery of RCAS-Myc producer cells stimulated tumor formation only in conjunction with tumor-suppressor gene deletion and only in a subset of injected mice (Lewis et al. 2003). Moreover, the low infection rate means that multiple viruses can be delivered to the same cell in vivo only where local virus delivery is possible (e.g., during intracranial injection into the brain [Holland et al. 1998a]), but not in instances where systemic virus delivery is used. Together, these weaknesses limit the specific candidate genes that can be effectively studied in specific tissues.
Tumor Models Using RCAS-TVA
The RCAS-TVA system has been used to generate models for many human cancers and, more recently, to target components of tumor stroma. The viral delivery process via the TVA receptor allows for many levels of control and customization. What follows is a brief summary of several models that have taken advantage of these possibilities.
The first in vivo experiment using the RCAS-TVA system used a transgenic mouse expressing the TVA receptor under the α-sk-actin promoter (Federspiel et al. 1994). This results in TVA expression specifically in skeletal muscle. When RCAS virus containing the alkaline phosphatase (AP) gene was injected intramuscularly, AP was detectable in muscle tissue 7 d postinjection. No AP-positive cells could be detected in nontransgenic mice injected with RCAS-AP (Federspiel et al. 1994). This experiment provided proof of principle for the use of the RCAS-TVA system to deliver genes in a postnatal and sporadic manner to mammalian cells in vivo.
The RCAS-TVA system was next used to model gliomas. GFAP-TVA transgenic mice were developed that specifically target RCAS infection to astrocytes. When DF1 cells producing RCAS-bFGF virus were intracranially injected into newborn mice, infected cells were found to have an increase in both proliferation and migration, but no gliomas formed in these mice (Holland and Varmus 1998). Gliomas did form, however, when a constitutively activated form of EGFR was delivered into GFAP-TVA mice with deletion at the Ink4a/Arf locus. Gliomas also formed when RCAS viruses encoding Cdk4 and activated EGFR were simultaneously injected. This model showed that EGFR activation could induce gliomas in cooperation with disrupted cell cycle arrest pathways (Holland et al. 1998a).
Holland and colleagues also used the Nestin gene promoter to drive TVA expression in glial progenitor cells (Holland et al. 1998b). To compare the sensitivity of different cell lineages to glioma formation, RCAS viruses containing activated Kras and Akt were delivered intracranially in either GFAP-TVA or Nestin-TVA mice. Although no tumors formed when activated Ras and Akt were expressed in the differentiated astrocyte lineage from the GFAP-TVA mice, Nestin-TVA mice developed high-grade gliomas (Holland et al. 2000). These data showed the impact of the differentiation state of the target on sensitivity to specific genetic alterations.
Studies using the Nestin-TVA strain have showed the importance of the initiating oncogenic lesion on the resulting tumor type. In human medulloblastoma, aberrant activation of hedgehog signaling commonly occurs. When RCAS-SHH was injected intracranially into Nestin-TVA mice, medulloblastomas, but not gliomas, formed in 9% of injected mice (Rao et al. 2003). Simultaneous delivery of other oncogenes, including Akt, which was shown to induce glioma development in cooperation with Kras, cooperated with SHH to induce medulloblastoma (Rao et al. 2003, 2004; Doucette et al. 2012). Thus, using a single transgenic mouse line and a collection of RCAS viruses, two very different tumor types were effectively modeled in vivo. A similar impact of the initiating oncogenic lesion on the resulting tumor type was observed in pancreatic cancer models generated using the Elastase-TVA transgenic line (Lewis et al. 2003; Morton et al. 2008).
Recently, Seidler and colleagues described a TVA mouse line that should have applicability across a broad range of tumor types. They generated a TVA transgenic line in which expression of TVA is regulated by the ubiquitous Rosa26 locus. In this mouse strain, a transcriptional stop cassette flanked by loxP sites precedes the TVA cDNA, preventing expression in the absence of the Cre recombinase (Soriano 1999). Breeding of this line to an appropriate Cre driver line results in targeted TVA expression and RCAS virus infection. Using a protamine-Cre line, which activates Cre in the germline, they showed that cells from a wide range of tissues could be infected following RCAS virus delivery (Seidler et al. 2008). The authors then combined this TVA line with the Ptf1a-Cre line that drives Cre expression specifically within the pancreas and showed that RCAS-Kras delivery stimulated the formation of pancreatic cancer precursor lesions. Tuveson and colleagues previously described a pancreatic cancer model in which activated KRASG12D is expressed from the endogenous locus (Hingorani et al. 2003). Seidler et al. combined their R26-TVA line with this model, and showed that delivery of RCAS viruses encoding a p53-targeting shRNA accelerate the progression to invasive carcinoma. Using RCAS-shRNA to target a tumor suppressor in vivo only affects a small percentage of cells, and thus more closely mimics the human condition versus other PDAC models where the entire pancreas has both activated Kras and mutation or loss of p53. Thus, the R26-TVA line can be effectively used to model tumor initiation and progression, and it should be a robust tool for the modeling of diverse tumors.
In addition to targeting cancer cells, RCAS-TVA models may also be used to model the impact of specific alterations on specific cell types that populate the tumor microenvironment. One such example is active endothelium, which forms blood vessels to support tumor formation. A mouse model expressing TVA under the Flk1 promoter results in TVA expression in proliferating endothelial cells (Vervoort et al. 2008). This results in TVA expression in the developing embryonic vasculature, subcutaneous implants in adult mice, and tumor-associated endothelial cells in adult mice. Limited infection of tumor-associated vasculature was showed when this TVA line was combined with the MMTV-PyMT mammary tumor model (Vervoort et al. 2008). This mouse model may be a useful tool in investigating gene perturbations that influence the function of tumor-associated vasculature.
In summary, RCAS-TVA mouse models of multiple human cancers have been generated, and these models have provided important insights into the cells of origin of particular tumors, and the roles of initiating genetic lesions in determining tumor type. The combination of RCAS-TVA models with existing transgenic, knockout, or knock-in models, and their deployment in screening studies, provides opportunities to extend the utility of these models.
ACKNOWLEDGMENTS
The authors apologize to colleagues whose work was not described in detail because of space limitations. Work in the Lewis laboratory is supported by grants from the National Institutes of Health.
- © 2014 Cold Spring Harbor Laboratory Press








