
Noninvasive Imaging of Tumor Burden and Molecular Pathways in Mouse Models of Cancer
- 1Lurie Family Imaging Center, Dana-Farber Cancer Institute, Boston, Massachusetts 02215;
- 2Department of Pathology, Harvard Medical School, Beth Israel Deaconess Medical Center, Boston, Massachusetts 02215;
- 3Department of Pediatrics, Columbia University Medical Center, New York, New York 10032
Abstract
Imaging plays a central role in the diagnosis of cancer and the evaluation of therapeutic efficacy in patients with cancer. Because macroscopic imaging is noninvasive and quantitative, the development of specialized instruments for small animals has spurred increasing utilization in preclinical cancer studies. Some small-animal imaging devices are miniaturized derivatives of clinical imaging modalities, including computed tomography, magnetic resonance imaging, positron-emission tomography, single-photon emission computed tomography, and ultrasonography. Optical imaging, including bioluminescence imaging and fluorescence imaging, has evolved from microscopic cellular imaging technologies. Here, we review how current imaging modalities are enabling high-resolution structural imaging with micrometer-scale spatial resolution, thus allowing for the quantification of tumor burden in genetically engineered and orthotopic models of cancer, where tumors develop within organs not typically accessible to measurements with calipers. Beyond measuring tumor size, imaging is increasingly being used to assess the activity of molecular pathways within tumors and to reveal the pharmacodynamic efficacy of targeted therapies. Each imaging technology has particular strengths and limitations, and we discuss how studies should be carefully designed to match the imaging approach to the primary experimental question.
INTRODUCTION
Over the past century, there have been tremendous advances in the technologies available for macroscopic imaging of humans and small animals. Beginning with the discovery of X-rays in 1895, advances in imaging technologies have improved the ability to visualize normal tissues and pathological lesions from the gross centimeter scale at the turn of the last century to micrometer resolution in the current era. Moreover, moving beyond anatomic imaging, the use of targeted probes and functional imaging methods now allows assessment of pathophysiology and normal physiology at the level of molecular pathways (Weissleder and Pittet 2008; Willmann et al. 2008b).
Imaging plays a crucial role in the diagnosis and staging of patients with cancer. Because imaging is noninvasive, quantitative, and readily amenable to longitudinal measurements, imaging is often the primary means by which the efficacy of cancer therapies is assessed in patients and in clinical trials. With the development of instrumentation specifically engineered for imaging small animals, the use of imaging in preclinical cancer research has grown rapidly in recent years.
The escalating demand for preclinical imaging has in part been driven by the need for preclinical models that better recapitulate human tumor biology (see Introduction: Translational Therapeutics in Genetically Engineered Mouse Models of Cancer [Olive and Politi 2014]). Long-standing concern about the predictive value of conventional subcutaneous xenograft models has spurred the increased use of orthotopic and genetically engineered mouse models (GEMMs) for cancer (Van Dyke and Jacks 2002; Olive and Tuveson 2006; Kung 2007; Talmadge et al. 2007). In these models, the development of disease within internal organs cannot be measured using calipers, as is commonly used for ectopic subcutaneous models. As in humans, the use of imaging in small-animal models allows for the noninvasive and longitudinal assessment of tumor burden, both to study the natural history of disease and to assess the impact of therapeutic interventions.
Beyond quantifying tumor burden, imaging is increasingly being used to assess the activity of molecular pathways and targets within tumors (Weissleder and Pittet 2008; Willmann et al. 2008b). The use of genetically encoded reporters or exogenous probes enables them to serve as biomarkers that can be noninvasively imaged to assess cellular phenotypes or the activity of molecular pathways. When applied to drug discovery, molecular imaging can be used to assess target engagement and modulation, thus establishing the pharmacodynamic efficacy of therapeutic interventions (i.e., establishing whether the drug modulates its target). The potential for molecular imaging to accelerate drug development has led to increased utilization in preclinical research, in part to validate these methods for potential use as early-response indicators in human clinical trials.
Overview of Small-Animal Imaging
Many small-animal imaging instruments are miniaturized derivatives of clinical imaging modalities, including computed tomography (CT), magnetic resonance imaging (MRI), positron-emission tomography (PET), single-photon emission CT (SPECT), and ultrasonography (US). Optical imaging techniques, including bioluminescence imaging (BLI) and fluorescence imaging, have evolved from microscopic cellular imaging technologies and currently are mostly restricted to preclinical utilization. Several prior reviews have detailed the principles of optical (Choy et al. 2003; Leblond et al. 2010), radionuclide (Shoghi 2009; Vanderheyden 2009), MRI (Vande Velde et al. 2009), and ultrasound imaging (Gessner and Dayton 2010). Here, we will focus on the practical aspects of imaging of mouse models of cancer.
Each imaging modality has specific strengths and limitations (described below and summarized in Table 1). When designing a preclinical study incorporating imaging, it is important to clearly define the biological question and then use the imaging modality that is optimally suited to achieve the desired end point. Under ideal circumstances, animal housing and imaging instruments will be colocalized within a barrier facility, thus allowing cross-modality imaging to interrogate different end points through the course of a longitudinal experiment.
Strengths and limitations of imaging approaches in animal models of cancer
There are some general considerations common to almost all imaging modalities. First, most imaging modalities require data acquisition over seconds to minutes of imaging, so mice need to be anesthetized for optimum image quality. Although injected anesthetics (e.g., ketamine/xylazine, Avertin) do not require additional equipment, the use of inhaled anesthetics (e.g., isoflurane, sevoflurane) is generally preferable based on animal-welfare considerations. The level of anesthesia can be titrated to optimize sedation using inhaled anesthetics delivered via a controlled nebulizer, which helps to prevent overdose. The effects of inhaled anesthetics are also rapidly reversed upon completion of imaging, by comparison with the fixed duration of the effects of injected anesthetics. These considerations are particularly important in sick tumor-bearing mice and where serial imaging is necessary (in some cases, hourly for pharmacodynamic studies).
Physiological monitoring and control are also key considerations. Maintenance of body temperature is crucial while animals are under sedation. Many instruments have heated stages or gurneys that are generally adequate for short periods of imaging. Prolonged imaging can necessitate core temperature monitoring (e.g., by rectal probe) for more precise temperature regulation. Cardiac and respiratory monitoring also provides important information about animal well-being and can be used for gated imaging (e.g., MRI, CT, US) to reduce motion artifacts for thoracic and abdominal imaging. Many instruments have integrated systems for physiological monitoring and support, and third-party options are also available, but compatibility with the imaging instrument must be ensured (e.g., MRI-safe). Temperature support during anesthesia induction and recovery is also important for sick tumor-bearing mice and can be accomplished by placing cages on circulating water pads. The use of chemical hot packs must be carefully vetted for possible hyperthermia and, where necessary, compatibility with MRI (iron powder is the primary ingredient in some heat packs).
PET, SPECT, and CT Imaging
PET, SPECT, and CT are common imaging modalities in both clinical and preclinical settings. Although CT scanning uses the differences in tissue X-ray photon attenuation to provide high-resolution anatomic images, nuclear imaging methods, such as PET and SPECT, use tracer quantities of radiolabeled probes to furnish functional and molecular information. For both clinical and preclinical imaging, PET and SPECT images are often coregistered with CT imaging to provide anatomic context for the nuclear imaging. This context is important because, although nuclear imaging approaches are highly sensitive and can detect picomolar concentrations of probe (Gambhir 2002), their spatial resolution is poor (1–2 mm). Moreover, the more specific the probe the less normal anatomy is visualized. PET–CT and SPECT–CT fused-modality instruments are now the norm for clinical and preclinical imaging, and trimodality PET–SPECT–CT instruments are also available in the preclinical setting.
The nuclear imaging methods all rely on administration of an exogenous radiolabeled probe. The most commonly used nuclear imaging probe is 2-deoxy-2-(18F)fluoro-d-glucose (18F-FDG), which is preferentially transported into and retained in cancer cells owing to their increased expression of glucose transporters and activation of the glycolytic pathway (Haberkorn et al. 1994). (See Protocol: 18F-FDG-PET/CT Imaging of Drug-Induced Metabolic Changes in Genetically Engineered Mouse Lung Cancer Models [Wang and Kung 2014].) In the clinic, FDG PET is primarily used for diagnostic purposes, but there is increasing interest in its use as a pharmacodynamic readout of drug activity. In preclinical and clinical studies, changes in FDG avidity can be observed in some cases after a single dose of targeted therapy (Van den Abbeele 2008). In these cases, an acute change (within hours or days) in FDG signal by PET imaging reflects the metabolic changes induced by target inhibition (i.e., the drug engages its target), rather than changes in tumor bulk, which might not be apparent for weeks or even months of treatment. Used in this manner, FDG can be considered a molecular imaging probe for tumor metabolism (Fig. 1).
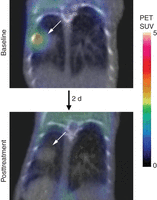
Imaging pharmacodynamic efficacy using FDG-PET. Baseline FDG-PET imaging reveals FDG avidity in tumors (arrows) arising in a genetically engineered mouse model of lung cancer driven by the L858R mutant epidermal growth factor receptor (EGFR). After 2 d of treatment with the irreversible EGFR inhibitor WZ4002, repeat FDG-PET imaging shows loss of FDG avidity. PET images (color scale) are combined with CT (gray scale) for anatomic reference, demonstrating no change in tumor size. These results show the utility of FDG-PET imaging to demonstrate rapidly pharmacodynamic efficacy (metabolic change) preceding any change in tumor volume. SUV, standardized uptake value.
Although thousands of nuclear imaging probes have been developed for research purposes, very few are currently clinically used for cancer imaging and readily available for preclinical imaging. In addition to FDG, other commonly available cancer imaging probes include (18F)-3′-fluoro-3′-deoxy-l-thymidine (18F-FLT) and 18F sodium fluoride (18F-NaF). Physiological tracers, such as 11C, 13N, and 15O, require an on-site cyclotron owing to the short half-lives of these species (20, 10, and 2 min, respectively) and are not readily available to the general research community.
CT imaging is based on the principle that tissues absorb X-rays to varying degrees. Accordingly, contrast is greatest when used to assess tissues with large differences in densities, such as bone compared with soft tissues (skeletal imaging) or air compared with tumor (lung tumor imaging). Although resolution down to ∼10 µm can be achieved with high-resolution CT imaging, this resolution requires significant imaging time, resulting in a considerable radiation dose. For serial imaging purposes, careful attention to dosimetry is necessary to keep doses below biologically active levels, which can necessitate a trade-off in terms of decreased spatial resolution. Serial high-resolution CT imaging can result in cumulative radiation doses that are sufficient to alter tumor growth.
MRI Imaging
MRI imaging is based on the polarization of protons in tissues when subjected to a strong magnetic field, with the subsequent emission of weak radio waves that can be detected during the relaxation phase. MRI imaging provides excellent spatial resolution (<10 µm) without the need for ionizing radiation and produces excellent tissue contrast that is superior to that of CT. MRI has been used for small-animal imaging for more than 30 years (Hansen et al. 1980; De Luca et al. 1982), with significant advances over the past decade resulting in the development of sophisticated high-resolution instruments specifically engineered for small-animal imaging. Small-animal MRIs were originally based on conventional nuclear magnetic resonance devices using vertical bores. Current small-animal MRIs have both vertical and horizontal bores, with horizontal bore instruments enabling easier manipulation and monitoring of animals.
As human organs are significantly larger than those of rodents, proportionately higher spatial resolution is necessary to achieve comparable anatomic detail when imaging mice. As such, although human MRI uses field strengths of 1.5 or 3 T, typical small-animal magnets use higher field strengths of 7, 9.4, or 11.7 T or even higher. Development of such high-field-strength instruments poses significant engineering challenges and significant hardware and space requirements. Furthermore, although high field strength increases the signal-to-noise parameters of MRI, it also introduces high-field imaging artifacts. Together, these challenges necessitate physics and engineering support for small-animal MRI setups, including development of optimized pulse sequences and design/construction of radio frequency (RF) coils to optimize signal to noise. (See Protocol: Preclinical Magnetic Resonance Imaging in Mouse Cancer Models [Sun and Kung 2014].)
The space and resource requirements for high-field-strength MRI are often prohibitive, spurring the recent development of low-field-strength “desktop” MRI instruments, including a 1-T self-shielded MRI scanner with compact dimensions and lower cost. This cryogen-free permanent-magnet MRI system can be used for routine animal imaging and has the potential for multimodality imaging applications, such as PET–MRI (Schmid et al. 2012). Given the low field strength, imaging times must be proportionally increased to achieve resolution comparable with that of higher field strength instruments.
MRI is most commonly used for structural imaging owing to the excellent soft-tissue contrast that can be achieved. Differences in spin-lattice or longitudinal relaxation (T1) and spin–spin or transverse relaxation (T2) provide tissue contrast and can be used to distinguish diseased tissue from normal tissue (Fig. 2). Prolonged T1 and T2 values are often associated with malignant tumors, and changes of T1 and T2 can be used as a biomarker for evaluation of treatment (Schad et al. 1989; Macri et al. 1992; Sun et al. 2004). In addition to T1 and T2 values, high-field MRIs can also be used to interrogate a variety of functional end points. MR spectroscopy can be used to investigate metabolite concentrations, local lipid content, and pH (Raghunand 2006). The full MR spectra of tissue can be used to establish a chemical fingerprint for characterizing the tumor type, tumor stage and tumor treatment response (Zoula et al. 2003).
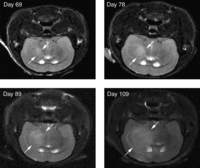
MRI of orthotopic brain tumors. Primary human glioblastoma multiforme cells were stereotactically implanted into the brains of mice. MRI imaging shows progressive growth of primary orthografts at the indicated time after implantation. The tumor (boundary indicated by arrows) is distinguishable from surrounding brain tissue on the basis of T2 hyperintensity (transverse relaxation).
Diffusion-weighted imaging is sensitive to the molecular movement of water and can be used to visualize changes in the diffusion of tissue water, thus providing a tool to noninvasively measure cellularity and differentiate tumor from normal tissues (Herneth et al. 2003). Apparent diffusion coefficient (ADC) measurements can be used to study the diffusion characteristics of tissue at a microscopic level. For example, brain tumors have significantly higher ADC values than normal brain tissue (Chenevert et al. 1997; Sun et al. 2004). Diffusion tensor imaging provides further information about the degree and direction of water diffusion within individual units of volume, and fiber-track reconstruction, or tractography, allows visualization of neural connections and fiber orientation from the direction of diffusion (Mori et al. 1999; Basser et al. 2000).
Perfusion MRI with methods of dynamic susceptibility MRI (DSC), dynamic contrast-enhanced MRI, and arterial spin labeling can be used to access vessel dilatation, blood vessel volume, and permeability, providing a tool to evaluate tumor angiogenesis and the effect of vascular-disrupting therapies (Barrett et al. 2007). Likewise, for vascular assessment, dynamic contrast-enhanced MRI (DCE-MRI) can be used to assess vascular volume (Jordan et al. 2005), and magnetic resonance angiography can be used to measure the degree of vessel tortuosity, vessel density distribution, and branching pattern (Bullitt et al. 2003; Fink et al. 2003). A full description of the technical bases of these complex MRI methods is beyond the scope of this discussion, but these technologies hold great promise as translatable functional imaging modalities.
Optical Imaging
BLI and fluorescence imaging in small animals is predicated on the principle that photons can pass through tissues. For BLI, light is produced by cells expressing a luciferase species. Fluorescence imaging uses excitation of genetically encoded fluorescent proteins or exogenously administered fluorophores to emit light. Absorption by tissues and hemoglobin significantly attenuates penetration of light with wavelengths <650 nm and so both BLI and fluorescence imaging are facilitated by light emission within the red or near-infrared regions of the spectrum.
BLI was initially developed for modeling infectious diseases, allowing bioluminescent bacteria to be imaged in live animals (Contag et al. 1995). The ability to engineer almost any cell type for BLI (Fig. 3) has prompted widespread adoption of bioluminescence imaging for cancer research applications (Contag et al. 1997; Prescher and Contag 2010). (See Protocol: Quantitative Bioluminescence Imaging of Mouse Tumor Models [Tseng and Kung 2014].) BLI necessitates that cells or animals be genetically engineered to express a variety of luciferase species, most commonly firefly luciferase. Injection of a cognate substrate (e.g., beetle d-luciferin for firefly luciferase) results in an enzyme-catalyzed oxidation reaction with the production of light. Because intrinsic bioluminescence is negligible in mammalian tissues, background is minimal, and thus BLI has outstanding signal-to-noise ratios and excellent sensitivity (Zhao et al. 2005) (see Introduction: Imaging Mouse Cancer Models In Vivo Using Reporter Transgenes [Lyons et al. 2013]).
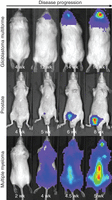
Bioluminescence imaging (BLI) of orthotopic tumors. Human primary glioblastoma multiforme cells, engineered prostate cancer cells, and multiple myeloma cells were engineered to express firefly luciferase. Orthotopic tumors were established by stereotactic implant into the brain, injection into the ventral prostate, or intravenous injection, respectively. Tumor burden was assessed by serial bioluminescence imaging. For each model, all images are represented at the same scale.
Fluorescence imaging can use genetically encoded fluorescent proteins or exogenous fluorophore-based probes. Although reagents based on green-fluorescent protein (GFP) are pervasive for in vitro studies, the excitation–emission properties of GFP are highly unfavorable for in vivo imaging applications, because of both strong tissue absorption and high tissue autofluorescence (Tam et al. 2007). Except for the imaging of superficial dermal lesions, GFP is generally unsuitable for in vivo imaging. Utilization of red-shifted fluorescent species (>650 nm) improves tissue penetration and lowers autofluorescence (Weissleder and Ntziachristos 2003; Leblond et al. 2010). Fluorescent probes can be used to assess the vascular pool, can be targeted to specific cell-surface proteins, or can be designed to change in fluorescent properties as physiological biosensors (Weissleder and Ntziachristos 2003).
Because of tissue scattering, the spatial resolution of macroscopic optical imaging is poor (>2 mm). Moreover, because light is strongly attenuated by depth, optical imaging techniques provide data that are relative in nature, rather than absolute. When used in a longitudinal manner, however, these data can be used in a quantitative manner, especially if correlated to established standards such as MRI tumor volume (Szentirmai et al. 2006). The difference in light scattering and attenuation at different wavelengths can be used to calculate the depth of a bioluminescence source. Because firefly luciferase has a broad emission spectrum, sequential planar images at defined wavelengths can be collected across the emission range (560–660 nm) and the signal intensity at each wavelength used to compute depth based on the known attenuation by tissue at each wavelength. This information can be used to reconstruct a three-dimensional (3D) projection from sequential planar images.
The major benefits of optical imaging modalities are ease of operation of the instruments, wide availability of reagents to enable imaging, high-throughput compared with MRI or nuclear-imaging approaches, and relatively less-expensive hardware. As applied to GEMMs, one impediment is the need to breed in conditional reporter alleles (Safran et al. 2003) that allow optical imaging of induced tumors. Given the complex genotypes of some GEMMs (three to four independent alleles), the addition of a reporter allele might be not only time consuming but could further decrease the fraction of weanlings with the appropriate genotype.
Ultrasound
Ultrasound imaging uses high-frequency sound waves that are reflected at frequencies based on echogenicity and captured by detectors. Echogenicity is influenced by several aspects of tissue composition, including water content. Fluids such as blood reflect less sound (are hypoechoic), whereas hard tissues such as bone as well as gas-filled tissues such as the lungs and gut reflect more sound (are hyperechoic). Thus, ultrasound is best suited for anatomical imaging of soft tissues and gas-free tissues, where the intrinsic properties of the tissue provide sufficient contrast to enable their identification.
Higher frequencies of ultrasound enable higher resolution imaging, but at the cost of decreased depth of penetration into tissues. For human patients, the need to image tissues at reasonable depths limits the usable frequency to ∼5 MHz for external transducers and ∼15 MHz for endoscopic transducers. However, high-frequency ultrasound transducers have been developed in the range of 25–75 MHz for use with small animals, where depth constraints are less of an issue. A typical 55-MHz mouse abdominal imaging transducer focuses in the 5- to 10-mm depth range, making it possible to visualize most anatomical structures at a resolution of 50 µm.
Ultrasound imaging does not use ionizing radiation and therefore can be performed serially without detriment. However, because US is a directed imaging technique, it has a limited field of view and cannot be used for whole-animal imaging. Although ultrasound images are two dimensional, 3D reconstructions can be achieved through mechanically driven stepwise acquisition of an image stack (Ayers et al. 2010), and 3D tomography can be used for accurate quantification of tumor volume. Because ultrasound can differentiate tissues based on intrinsic characteristics and does not require a genetically encoded reporter, it is well suited for use in tumor surveillance and therapeutic assessment in GEMMs (Cook et al. 2008).
In addition to anatomic imaging, ultrasound imaging can be used to assess blood flow and angiogenesis using Doppler mode imaging (Goertz et al. 2002). In recent years, the development of microbubble contrast agents has enhanced the utility of ultrasound to image vascular physiology; however, the ability to enhance cellular compartments outside of the vascular system remains limited. Microbubble contrast agents have been modified further with antibodies that can be used to detect and quantify the expression of antigens expressed on the luminal surface of vascular endothelial cells (Willmann 2008a). Complex microbubbles that encapsulate both a liquid and gaseous phase have been used for targeted delivery of drugs, thereby directly linking imaging and therapy. Advanced forms of functional ultrasound have also been developed that enable the measurement of tissue stiffness (ultrasound elastography and harmonic motion imaging) or that can even be used to burn tissues noninvasively in a focused manner, at depth (Maleke and Konofagou 2010; Maleke et al. 2010).
Ultrasound imaging can be used as a relatively high-throughput approach for tumor surveillance in GEMMs where tumor development is asynchronous and can span a several-month window. For example, methods for ultrasound imaging of GEMMs of pancreatic cancer have recently been described (Sastra and Olive 2013)—both for the purpose of identifying animals with established tumors to enroll on study as well as to monitor the effects of experimental therapeutics.
Experimental Workflow
A complete preclinical imaging workflow usually involves three stages.
-
1. Planning
-
Define the experimental question to be answered.
-
Pick the appropriate cancer model for study.
-
Determine the appropriate imaging modality and contrast agent/probe.
-
Design a comprehensive study, including time line, drug dose, route of administration, imaging points, secondary end points, and samples to be collected upon termination.
-
Identify mice with established tumors of adequate size.
-
-
2. Imaging
-
Prepare animals for imaging.
-
Inject contrast agents or probes.
-
Induce anesthesia.
-
Document experimental conditions.
-
Commence scanning and collection of raw data using system- and study-specific acquisition parameters.
-
Let animal recover postimaging.
-
-
3. Analysis
-
Conduct postprocessing of the raw data (e.g., tomographic image reconstruction).
-
Optimize display and interpretation of the reconstructed images.
-
Extract quantitative data from the images (e.g., quantitation of signal within specified regions of interest).
-
Use statistical analysis.
-
Use standardized storage systems and archive the raw and processed data.
-
All three stages are interdependent and should not be performed in isolation. For example, executing a sophisticated imaging procedure without careful consideration of the primary biological question might result in beautiful images that are entirely irrelevant. Analysis of data without knowing the imaging details and the questions to be answered could lead to incorrect interpretations. Furthermore, because of the necessity for using radiation in some modalities, there will be a trade-off between image quality and radiation exposure to the animals, which must be carefully balanced based on the overall study design (e.g., number of imaging points). Finally, while imaging studies can involve multiple personnel, this interdependency dictates that the best results will be achieved through a collaborative team approach.
In the associated protocols, we first describe FDG–PET imaging of early tumor metabolic changes induced by drug treatment, allowing the assessment of pharmacodynamic efficiency (see Protocol: 18F-FDG-PET/CT Imaging of Drug-Induced Metabolic Changes in Genetically Engineered Mouse Lung Cancer Models [Wang and Kung 2014]). Next, we show how to take advantage of the higher field strength of dedicated preclinical systems to conduct MR imaging with high spatial resolution (see Protocol: Preclinical Magnetic Resonance Imaging in Mouse Cancer Models [Sun and Kung 2014]). Finally, we describe and discuss the methodology for conducting BLI in genetically engineered mice (see Protocol: Quantitative Bioluminescence Imaging of Mouse Tumor Models [Tseng and Kung 2014]).
ACKNOWLEDGMENTS
The study was supported by the Nancy Lurie Marks Family Foundation.
- © 2015 Cold Spring Harbor Laboratory Press








