
Online Resources for Genomic Analysis Using High-Throughput Sequencing
- 1Department of Biochemistry and Molecular Biology, Penn State University, University Park, Pennsylvania 16802;
- 2Departments of Biology and Computer Science, Johns Hopkins University, Baltimore, Maryland 21211
Abstract
The availability of high-throughput sequencing has created enormous possibilities for scientific discovery. However, the massive amount of data being generated has resulted in a severe informatics bottleneck. A large number of tools exist for analyzing next-generation sequencing (NGS) data, yet often there remains a disconnect between these research tools and the ability of many researchers to use them. As a consequence, several online resources and communities have been developed to assist researchers with both the management and the analysis of sequencing data sets. Here we describe the use and applications of common file formats for coding and storing genomic data, consider several web-accessible open-source resources for the visualization and analysis of NGS data, and provide examples of typical analyses with links to further detailed exercises.
INTRODUCTION AND BACKGROUND
With the recent development and rapid proliferation of technologies for high-throughput sequencing (HTS)—also commonly called next-generation sequencing (NGS)—the generation of raw data is no longer a rate-limiting factor in many genome-wide studies. Experimental design and sample collection are certainly challenging, but data analysis continues to represent a formidable barrier for the majority of biomedical researchers. In fact, the scale of the data presents not only difficulties for individual researchers attempting analysis, but also significant informatics issues for collaboration, reproducibility, and dissemination. The basic requirements for conducting NGS analysis do not differ significantly from other research studies; in all these studies, raw data and results must be stored and shared, and the parameters for each step in the analysis methods must be tracked. In the complex analysis of NGS data, however, the magnitude of the data and the number of steps, as well as the parameters within each step, require a standardized strategy to ensure the analysis is a reproducible analysis.
The use of a standardized strategy is not meant to stifle scientific innovation by limiting the adoption and integration of newly developed algorithms, methods or technology. Instead, the strategy should encourage flexibility by constructing the analysis in a modular fashion. In this scheme, individual steps can be refactored or replaced with different approaches and algorithms as appropriate, and the individual steps and sets of steps should themselves be reusable. To a large extent, flexibility and reusability can be mediated through the adoption of and reliance on well-specified standard formats. However, an integrated approach is required to preserve parameter settings and intermediate data sets for the entire analysis pipeline along with the final results. Several online resources as well as software products are available to assist researchers with the management and analysis of sequencing data sets.
Online Tools
Although many closed source software products, commercial or otherwise, have been used successfully to advance science, these products are, by definition, incompatible with transparency and reproducibility. The reliance on such “black box” approaches does not adhere to the principles of open exchange of knowledge and materials which form the basis of scientific progress, and in fact has resulted in errors for a number of research groups (see e.g., Morin et al. 2012; Nekrutenko and Taylor 2012). Furthermore, many closed source software packages come with restrictive licensing terms and are cost prohibitive. For these reasons, in this introduction we focus on the open source software tools currently available for working with NGS data.
The growing collection of available online tools allows researchers to explore and analyze their sequencing data. Some of these, such as the UCSC Genome Browser (Kent et al. 2002) and the integrative genomics viewer (IGV; [Robinson et al. 2011]), are directed toward enabling visualization of genomic data. Other tools, such as Galaxy (Giardine et al. 2005; Blankenberg et al. 2010b; Goecks et al. 2010), the Genomic HyperBrowser (Sandve et al. 2010), the BioExtract Server (Lushbough et al. 2011), GeneProf (Halbritter et al. 2011), Mobyle (Néron et al. 2009), and GenePattern (Reich et al. 2006), are focused on making stand-alone computational tools accessible and reproducible to biologists. We consider these tools and their functionality in more detail in Online Analysis Tools.
Online Support Forums
When confronted with analyzing a set of data, studying the primary literature—although necessary and important—is no longer sufficient for determining the best course of action to follow. Best practice approaches are quickly evolving, with new tools and new versions being created at breakneck speed. Even the smallest change in a parameter setting can have a profound impact on the final conclusions of a study. Many online manuals for tools are incomplete, and—even when functionally complete—often do not fully describe the implications of individual parameter settings on the internal behavior and resultant output. Confounding issues include the interplay among individual parameters and the consideration that preferred settings for a particular technology may not have existed at the time of its development. To assist users beyond static manuals, most active software projects provide support through the use of mailing-lists or dedicated forums. But these support avenues are often staffed only by the tool developers who, despite their best efforts, may be delayed in responding and/or limited to helping only with minor issues such as getting the software to compile or run.
Fortunately, a number of knowledgeable independent communities have sprung up across the Internet, such as SEQanswers (Li et al. 2012) and BioStar (Parnell et al. 2011). SEQanswers, launched in 2007, takes the approach of an open forum—anyone can create an account and users are encouraged to initiate and participate in threaded discussions. This forum has facilitated not only the evaluation of current analysis standards, but also the development of new techniques and analysis methods. BioStar is modeled on the question and answer website Stack Exchange (http://stackexchange.com), where users are subject to a “reputation award” process. In this forum, a participant asks a specific question and site users provide direct answers. Other participants then vote on each answer provided, and the questioner is given the option of approving one or more answers (typically the answer(s) that seemed most appropriate or useful). The answers with the most votes are ranked and rise to the top of the page (in contrast to the approach in a timestamp-based forum). This system has the advantage of providing concise answers to specific questions, which can be easier for users to find and follow. Participants are granted “reputation” points and awards based on the community assessment of their contributions—an application of gamification to encourage user engagement.
DATA FORMATS AND USAGE
Before elaborating on the functionality of online analytic tools, we first explore topics relevant to the representation of most common types of genomic data. One of the first barriers encountered by researchers working with NGS data sets is the data itself. The individual stages of any particular analysis have different information that must be encoded and stored as electronic files. As well as the various types of information that must be stored (e.g., raw sequencing reads, aligned sequencing reads, genome assemblies, called variants, genomic regions, etc.), there exists also a plethora of different formats available for each type of information. To impose some consistency, the research community has adopted a few of these formats as standards. It should be noted that, as with the steps of a preferred analysis pipeline, the reliance on any particular format is likely to change over time. The formats we describe here (see also Table 1) are representative of the common file formats currently in use; all of these formats are open source with publicly available specifications.
Commonly used file formats
Sequencing Reads
A sequencing read is the functional unit of generally usable information that is the output of sequencing strands of nucleic acids. The FASTQ format has become the de facto standard format for the representation of sequencing reads (Cock et al. 2010). FASTQ files are a plaintext format that contains a read identifier, the sequenced nucleotide bases (i.e., A, T, G, C) and a quality score for each base, indicating the probability of each base being called correctly. Confusingly, there are several different FASTQ variants. The preferred format is the Sanger variant which relies on the use of Phred quality scores, the widely accepted convention for calculating and depicting the quality of a sequence. In the Sanger FASTQ variant, the quality scores are Phred-scaled and encoded using ASCII characters. Each character indicates the quality of a specific sequenced base, where the Phred-scaled value is the ordinal value of that character subtracted by 33.
Alignments Against a Reference
A common step following sequencing is the alignment of the sequencing reads to a reference genome or transcriptome; this process is also known as “mapping reads” (Trapnell and Salzberg 2009). The preferred format for representing mapped reads is the SAM/BAM format (Li et al. 2009). The SAM format is a human-readable, tab-delimited flat (plaintext) file containing a great deal of information about the read mapping. These features include, among others, the name of the read, the read sequence, base quality scores, the mapped chromosome and position within the reference sequence, mapping quality, CIGAR string (a representation of the alignment characteristics of each base in the read). The BAM format is a block-compressed binary representation of the same data contained within the SAM format; it is generally much smaller in size than the plaintext equivalent. Conversion between the two representations is straightforward; several conversion programs are available and may be required, depending on the formats accepted by any particular tool (a tool may accept only SAM, or only BAM, or both). The SAM/BAM format has been designed to be extensible through the use of user-defined “tags.” In addition to the BAM format, a reference-based compression format, known as CRAM (http://www.ebi.ac.uk/ena/about/cram_toolkit), has also been developed. CRAM is able to perform file compression with a purported space efficiency increase of up to approximately twofold, typically in one of two ways, by lossless or by lossy compression. In files compressed by lossless compression, all of the data information originally present in the file is completely recovered (restored) when the file is uncompressed; whereas lossy compression is accomplished by eliminating part of the file information, and that information remains permanently lost on decompression.
Variants
The goal of resequencing studies that follow mapping reads to a reference genome is often to determine the differences between the reference and the sequenced reads. The variant call format (VCF; Danecek et al. 2011) is a tab-delimited plaintext file designed to store sequence variation information—including single nucleotide polymorphisms (SNPs), insertions and deletions (indels), and larger structural variants—across any number of samples. Additional information for each variant beyond the standard specification can be defined using custom tags. This format also can be optionally compressed and indexed for increased data access and storage efficiency.
Genomic Regions
Among the multitude of available formats used to encode genomic region and feature information, the two most popular are the browser extensible data (BED; https://genome.ucsc.edu/FAQ/FAQformat.html) and the generic feature format (GFF; http://www.sequenceontology.org/gff3.shtml). Both formats are tab-delimited plaintext, however they have several important differences, the most significant of which is the use of different coordinate systems. BED files can exist in various configurations containing between 3 and 12 columns, and represent zero-based half-open intervals; the first base is 0 and the position indicated in the value of the end fields is not included (the length of a feature can be calculated as end – start). GFF, on the other hand, is one-based, fully closed; the first base is 1 and the end position is included in the interval (the length of a feature is generally calculated as end – start+1). There are several versions of the GFF format; at the time of this writing, version 3 of the specification is the latest. When working with genomic region information, it is important not to mix different coordinate systems inadvertently, without taking the differences into account. Inconsistencies can easily arise—the most common of which is an off-by-one error.
GENOME ASSEMBLY
Genome assembly is the process of generating a set of longer contiguous sequences from the original collection of sequencing reads, with the goal of reconstructing the sequence of the original source chromosomes. This assembly can be achieved either in a de novo manner or by using a preexisting reference genome as a backbone. The finalized version of an assembled genome is often made available in the FASTA format. This format is composed of blocks of a header line demarcated with a greater than sign (>) and followed by lines containing the sequence represented by single character codes representing nucleotide bases; FASTA files can also store peptide sequences. A FASTA file can contain any number of FASTA blocks, where each FASTA block begins with its own header line and may or may not be separated by an empty line. Other formats can also be used. The 2bit format, for example, is a randomly accessible binary format that contains the names and DNA sequences, including masking information. Masking is a process often used to identify and remove repetitive and low-complexity sequences to prevent them from being used within, for example, alignment procedures.
Genome Builds
Each version of a genome assembly is commonly known as a “genome build.” It is of particular importance to understand that there are significant differences between genome builds, even when they derive from the same organism or set of individuals. These variations may occur, for example, as different coordinates or as differing sequence content for various sequence elements. The exact results of an analysis performed using one genome build cannot be assumed to be valid for another genome build, however, the general conclusions of an analysis often remain valid. Great care should be taken to ensure that genome builds are not intermixed accidentally, for example, mixing a BED file from one genome build with a FASTA file from another.
Moving between Genome Builds
Moving a set of coordinates from one genome build to another is a process known as remapping or liftover. In this approach, an alignment between the genomes is computed. Using the coordinates of the aligning genomic segments, it is possible to convert coordinates between genome builds and across species. Two popular online tools for this process are the NCBI Genome Remapping Service (http://www.ncbi.nlm.nih.gov/genome/tools/remap) and the UCSC Liftover tool (http://genome.ucsc.edu/cgi-bin/hgLiftOver). UCSC Liftover allows moving between different species, for example, identifying the corresponding coordinates of a gene of interest in another species. However, Liftover is not a substitute for de novo annotation. Extra caution should be taken when using this process, particularly when moving such a set of coordinates between species. Underlying assumptions about the accuracy of features in the corresponding regions increase in uncertainty as the distance between the genome builds increases. A command-line version of the UCSC Liftover tool can also be downloaded for local use, and has been incorporated into other online tools, such as Galaxy, as we describe below.
GENOME BROWSERS AND VISUALIZATIONS
An ever-growing collection of genomic data is becoming publicly available. One of the most effective ways to examine this data is through visualization. Genome browsers provide a graphical depiction of biological database information, in which one axis (commonly the x-axis) represents the location along the genome (i.e., genome coordinates) with the free space above this axis occupied by data from several different “tracks” corresponding to a gene or a coordinate. These tracks typically include a sequence track (i.e., the nucleotide bases) as well as a varying array of different annotation tracks that may provide gene predictions, comparative analysis, gene regulation, gene expression, etc. Each of these individual tracks typically occupies its own subsections of the y-axis, and each subsection may have its own y-axis scale for displaying conservation or other scores. To denote gene predictions, the structures of the predicted genes (i.e., exons, introns, UTRs, etc.) are represented using graphical icons, sometimes referred to as “glyphs.” We consider here three widely used browsers, each offering particular features and advantages.
UCSC Genome Browser
The UCSC Bioinformatics group has developed many tools and resources for the genomic community, notably the UCSC Genome Browser (Kent et al. 2002) and the UCSC Table Browser (Karolchik et al. 2004). The Genome Browser allows users to visualize preloaded genomic annotation tracks as well as their own data tracks. The Table Browser allows downloads of the data tracks presented within the Genome Browser, either in an unmodified (unfiltered) format or after applying various filters, intersections, or transformations. Data can also be exported directly to external resources such as Galaxy. The UCSC Bioinformatics group also provides access to a public MySQL server that contains the same data available from the Genome Browser.
The primary public UCSC Genome Browser is focused on vertebrate species (as well as a few other model organisms) and is located at http://genome.ucsc.edu/cgi-bin/hgGateway, with several mirror sites available across the globe. Genome Browsers that focus on other species groups, such as the Archaeal Genome Browser at http://archaea.ucsc.edu/ (Schneider et al. 2006), are also available. The entry or gateway page of the Genome Browser allows the user to select the clade, species, and genome build of interest. Once the desired genome build has been selected, the user can enter a query within the “search term” box and click “submit” to jump to the corresponding location within the annotation tracks page. When a query term matches several locations, the user is presented with a selectable list of matching locations. Several types of queries may be considered, including chromosomal position ranges or bands, gene symbols, accession numbers of mRNA and ESTs, and descriptive terms that are found within GenBank mRNA records. If a user has genomic DNA, mRNA, or protein sequence, but does not know valid name or location, the online BLAT tool (Kent 2002) can be used to create a report of homologous positions that will contain links for viewing the selected alignment within the Genome Browser. Several external web applications—Galaxy, Entrez Gene (Maglott et al. 2005), AceView (Thierry-Mieg and Thierry-Mieg 2006), Ensembl (Flicek et al. 2013), SUPERFAMILY (Gough et al. 2001), and GeneCards (Safran et al. 2010)—also provide direct links to Genome Browser positions.
The Genome Browser provides the ability for users to upload their own data for use as custom tracks. Custom data can be uploaded by external applications, individually by the user or through a system known as Track Data Hubs. Track hubs are sets of described directories containing genomic data that can be public or private. Track hubs allow the efficient creation of large customizable browser tracks that have the same functionality as built-in tracks including grouping as composite or supertracks.
If an annotated reference genome is not available at the Genome Browser, users can take advantage of the Assembly Hub functionality. Assembly Hubs are similar to track hubs, but here the users must also provide the underlying reference genome in the 2 bit compression format. This format allows users to harness the capabilities of the Genome Browser on nonstandard genomes, without having to run their own Genome Browser site, and by hosting only the necessary data in any standard webserver.
The Session Tool of the Genome Browser facilitates saving custom tracks, track views and other information between access times. A registered account user can save multiple sessions, allowing one user to work on multiple tasks without interfering with another. Sessions can be saved, loaded, deleted, and shared. A user who has customized the browser view and would like to create a screenshot (for example, for inclusion in a manuscript) can access the PDF/PS option under the View menu in the top blue bar of the Genome Browser. Here, the user can export the current annotation track view or the chromosome ideogram in either PDF or EPS format.
Ensembl
The Ensembl project (Flicek et al. 2013), a collaboration between the European Bioinformatics Institute and the Wellcome Trust Sanger Institute, provides the free web-based Ensembl Genome Browser at http://www.ensembl.org. This genome browser is focused on providing access to fully sequenced vertebrate and selected eukaryotic model organisms. A sister project, EnsemblGenomes (http://ensemblgenomes.org/), has been developed to extend access to nonvertebrate genomes. Ensembl uses an automated pipeline to annotate genomes, which are stored in a set of core databases. These databases can be accessed visually using the genome browser, interactively explored using BioMart, or queried using software known as an application programming interface (API).
The Ensembl browser allows users to visualize public data sets along with uploaded custom data tracks. Users can add custom data by uploading or providing the URL of properly formatted files and by accessing a DAS server, or they can optionally disable the display of data tracks that are not of interest to their research. In addition to the classic genome browser display, additional views are available, including a synteny display, a gene view, a transcript view, and resequencing data tracks view. The Ensembl Genome Browser provides a user registration system that allows bookmarks to be created, custom data tracks to be saved between browser sessions, and track configurations to be saved.
Integrative Genomics Browser (IGV)
The Integrative Genomics Browser (IGV) (Robinson et al. 2011) is a Java-based visualization tool for genomic sequence and annotation data. Two versions are available —one can be downloaded and the other (a web-start version) can be launched from within a web-browser or via a shared URL. By making use of several indexing strategies, on-demand data loading, and a specialized binary multiresolution tiled data format, IGV supports viewing a large amount of data for a wide range of data formats, including those from array-based and next-generation sequencing studies along with genome annotations. IGV includes a “multilocus” mode that enables viewing multiple noncontiguous genomic regions within the same window.
IGV offers a default set of built-in data for several genome builds, including genomic sequences, chromosome ideograms and reference gene tracks. Custom genome data can be specified for nonbuilt-in genome builds, and additional data can be loaded for display as annotation tracks. Data can be loaded into IGV by using any of a number of approaches—uploading from the user's computer, entering a web-accessible URL containing the data, by accessing a distributed annotation system (DAS) source, or by loading from the IGV server.
ONLINE ANALYSIS TOOLS
Galaxy
Galaxy (Giardine et al. 2005; Blankenberg et al. 2010b; Goecks et al. 2010) is an open, web-based platform for accessible, reproducible, and transparent computational biomedical research. Galaxy makes bioinformatics analyses accessible to users lacking programming experience by enabling them to easily specify parameters for running tools and workflows. Analyses are made transparent by allowing users simple access to share and publish analyses via the web and create Pages—interactive, web-based documents that describe a complete analysis. Figure 1 provides an overview of Galaxy's Analyze Data interface.
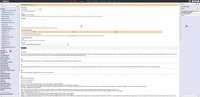
The Galaxy analysis interface. The Galaxy analysis interface is constructed of four main parts: (A) the masthead at the top, (B) the tools menu on the left, (C) the tool interface in the center, and (D) the analysis History located on the right. Here, the Upload tool interface is visible after being selected from the “Get Data” section of the tools menu. The analysis interface is the default Galaxy view and can be accessed using the Analyze Data link (E) from within the masthead.
A free public instance of Galaxy is available at http://usegalaxy.org and additional help for using Galaxy beyond that provided here can be found at http://galaxyproject.org. In addition to this introduction, users who are not familiar with Galaxy are directed to follow the tutorial available at http://usegalaxy.org/galaxy101. Signing up for a user account is optional but recommended as many of Galaxy's advanced functions, such as saving multiple Histories or editing workflows, require that the user be logged in. Registered account users also have access to larger disk usage quotas than do nonregistered anonymous users.
The History system is at the heart of the reproducibility and provenance provided by Galaxy. When a tool is run in Galaxy, it creates one or more output data sets to be placed into the user's History. As an analysis is interactively performed within Galaxy, the outputs of each tool are stored with comprehensive information about the running of each job, including the selected input data sets (if any) and the values of each parameter used within the particular tool execution. Thus, the History is a perpetual container for the input and output data sets of any analysis tool. By default, many Galaxy tools are configured with a set of best-guess default parameters. However, relying on the default settings is not always the best course of action, particularly for a complex NGS analysis. Often, the most useful and relevant parameters are exposed within the default tool configuration view, but using the advanced parameters widget provides access to the complete complement of tool parameters.
Galaxy Data Sets
Data sets are the inputs and outputs of analysis jobs and are the focal point for much of the power of Galaxy. To ensure reproducibility, Galaxy data sets are immutable objects, that is, once created the data content cannot be modified. Data sets can be loaded into a History in a number of ways—by uploading from the user's computer; fetching from a provided URL; pasting content into a textbox; importing from a Data Library, shared History, or Galaxy Page; or as a result of an analysis or data source tool (Blankenberg et al. 2011).
Additional actions can be performed on data sets depending on the datatype and metadata. For example, BED or BAM files belonging to certain genome builds may be viewed at resources external to Galaxy such as the UCSC Genome Browser, Gbrowse (Stein et al. 2002), or IGV. These external resources appear as links within the expanded data set. Several resources are included with Galaxy by default and the administrator of the Galaxy instance can add new external links using a plugin system.
By using the rerun button, the user can automatically populate the middle tool interface with the tool, input data sets, and parameter settings that were originally used in the analysis. The user can then choose either to repeat the analysis step according to the original settings or to change the values of any of the tool settings before reexecuting the analysis step. In this execution, individual analysis steps can be rerun, or an entire analysis pipeline can be built automatically from a History by using the “Extract workflow” option from the History menu. Galaxy therefore simplifies the creation of a reusable analysis from an interactively created series of analysis steps; because all the information needed to create the workflow is automatically stored as an inherent property of Galaxy's tool framework, no additional effort is required by the user to indicate that the system should start to record the steps being performed. Whereas Galaxy workflows can be created automatically from a previously performed analysis, workflows can also be created and edited interactively using a drag and drop graphical interface (Fig. 2).
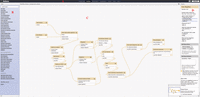
The Galaxy workflow editor. The workflow editor works with all standards-compliant modern web browsers and is composed of four sections: (A) the masthead, (B) the tools menu interface in the left-hand pane, (C) the workflow configuration canvas in the middle pane, and (D) the tool configuration interface in the right-hand pane.
Sharing Outcomes
Just as important as reproducibly performing a particular research study is the ability to effectively share the results and steps undertaken. Galaxy provides several facilities for sharing the outcome, steps, initial data, and methods write-up for any project. Essentially any Galaxy item can be shared at the discretion of its owner; these include individual data sets shared directly or through a Data Library, entire analysis Histories, visualizations (Goecks et al. 2012), and workflows. Galaxy items can be shared directly with another user by E-mail or with any number of target users by creating a link that allows access to any user who knows it. Finally, Galaxy items can be published to make them completely public, appearing in their respective lists under the Shared Data masthead menu. When sharing data sets directly or through libraries, Galaxy provides a role-based access control (RBAC) system that supports customized permissions through individual roles or through the use of user groups.
Galaxy Pages provide users with the ability to create documentation with a visual word-processing style editor to describe external experimental methods and any set of Galaxy items, including the rationale behind a particular analysis. These Pages have been proven effective by providing a complete overview of an analysis that serves as a “live supplement” to published manuscripts (e.g., Kosakovsky Pond et al. 2009) or as the basis for providing interactive tutorials. Within a given Page, links to designated Galaxy items can be provided or items can be directly embedded, allowing interaction with Histories, Data sets, workflows, and visualizations as well as importing for modification by any Galaxy user who can access the Page.
Several Galaxy instances are available for use free of charge, including the public instance provided by the Galaxy Team at http://usegalaxy.org, however, there may be a limited number of tools or insufficient disk usage quotas for a particular analysis. Fortunately, running a local instance of Galaxy on user-provided hardware is straightforward and extensively documented (http://getgalaxy.org). When a user lacks IT knowledge or access to adequate hardware, private Galaxy instances can be launched interactively through a web interface within commercial Cloud resources such as Amazon's EC2 (Afgan et al. 2010; see also http://usegalaxy.org/cloud). The Galaxy ToolShed (Blankenberg et al. 2014; http://usegalaxy.org/toolshed) provides a graphical interface for administrators to use in installing tools, dependencies, and other utilities into their own Galaxy instances that are not available by default.
A Typical NGS Analysis with Galaxy
A typical NGS analysis with Galaxy begins with loading sequencing reads in the FASTQ format into the History, either by uploading or by importing from an external data source such as the ENA Short Read Archive (Leinonen et al. 2011). After sequencing reads are loaded into Galaxy they can be analyzed for quality with the FastQC tool (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The reads are then filtered, trimmed, and/or otherwise manipulated as needed with the collection of tools located under the NGS: QC and Manipulation tool section (Blankenberg et al. 2010a). Except in cases of de novo assembly, the next step is to align the sequencing reads to a reference genome. When dealing with genomic DNA sequencing reads, the currently preferred mappers available within Galaxy are BWA (Li and Durbin 2009), Bowtie (Langmead et al. 2009), or, for longer reads, LASTZ (Harris 2007). When dealing with sequencing reads of an RNA origin (RNA-seq), a splice-junction mapper such as TopHat (Trapnell et al. 2009) should be used. Each of these tools will create SAM/BAM output that can be further analyzed.
Following alignment, SAMtools and Picard Tools (http://picard.sourceforge.net/) can be used to filter and manipulate the aligned sequencing reads. The next steps depend entirely on the type of experiment that was performed, such as ChIP-seq, variant detection, or RNA-seq. ChIP-seq experiments would require the use of peak or region callers, such as MACS (Zhang et al. 2008) or SICER (Zang et al. 2009), to find regions of the genome that are enriched for the mapped sequenced reads as identified by protein binding or histone modifications. A ChIP-seq exercise can be found at https://main.g2.bx.psu.edu/u/james/p/exercise-chip-seq. Variant detection and genotyping can be performed using tools such as FreeBayes (Garrison and Marth 2012), SAMtools mpileup, or the GATK (DePristo et al. 2011). RNA-seq analysis can be performed using the Cufflinks tool suite (Trapnell et al. 2010) and eXpress (http://bio.math.berkeley.edu/eXpress/). An RNA-seq exercise can be found at http://usegalaxy.org/rna-seq. Additional exercises, covering a wide range of topics, can be found under the Published Pages section (http://usegalaxy.org/page/list_published) of the Shared Data menu within the masthead of the main public Galaxy instance, and further step-by-step protocols are available in the literature (e.g., Hillman-Jackson et al. 2012).
When working with NGS tools within Galaxy it is of particular importance to take note of the reference genome that may need to be specified at several steps. Many Galaxy tools provide the user with the option to use either common built-in reference genomes or a user-provided reference genome (e.g., via a FASTA file in the user's History). When available, we recommend using a built-in reference genome, as these typically are preformatted to work with that particular tool (e.g., mapper index files). When, instead, a reference genome is selected from the user's History, it may be necessary at each individual step to automatically create one-off indexes for the genome provided, resulting in a process that is less efficient and can be quite time consuming.
The Genomic HyperBrowser
The Genomic HyperBrowser (Sandve et al. 2010) is a web-based statistical analysis system for genomic data that is integrated within a specialized version of the Galaxy framework. The HyperBrowser focuses on comparing two sets of genomic annotations to determine deviation from a null model. Here, genomic data sets are identified as one of five different types: (1) features occurring at specific base-pairs, known as points (unmarked points: UP); (2) features that span regions of a genome, known as segments (unmarked segments: US); (3) functions, where a value is assigned to each base pair (F), (4) valued points (marked points: MP); and (5) valued segments (marked segments: MS). Annotation tracks are selected either from a large list of built-in tracks or from tracks provided by the user via their current History. Once two annotations are selected, the user is presented with a predefined list of questions that varies based on the two types of data sets selected. The next step is to choose the null model that is most representative of the random events that characterize the two data sets. Based on the chosen null model and the question, the system then selects the appropriate statistical test, which may be either an exact test or a test based on a Monte Carlo approach. Results are returned either globally across the entire genome or for a set of bins with P-values or effect sizes calculated locally.
BioExtract Server
The BioExtract server (https://www.bioextract.org) is a free web-based service for designing and executing bioinformatics workflows. It provides access to hundreds of tools and data sources. Users are able to query and retrieve data sets from NCBI, EMBL, UniProt, and several plant-specific databases. These search results can be saved, filtered further, and used as input into analysis tools. Preexisting workflows can be executed, created by recording user steps, exported, and imported. BioExtract Server workflows have also been incorporated into myExperiment, a collaborative site and wiki that enables users to publish and share workflows and other digital objects.
CONCLUDING REMARKS
There is an ever-growing continually expanding collection of online resources available for visualizing and analyzing NGS data. Generally speaking, there is no perfect tool and each resource has its own set of advantages and drawbacks, however, it is in the interest of the researcher to determine the best tool currently available for a particular analysis. The NGS research space is undergoing rapid and continual development; therefore, because a particular resource was the best choice at a previous time does not mean it remains the best approach. In addition to the help available from individual tool developers and projects, researchers are advised to seek out assistance from community resources, such as BioStar and SeqAnswers, to inquire about the set of current best-practice tools and their usage, before making a serious start. If you have tried to search for an answer but are unsure about a particular resource, tool, or parameter, do not be afraid to reach out and ask a question—the online community genuinely wants to help.
ACKNOWLEDGMENTS
We, the investigators of this introduction, are lead members of the Galaxy Project team. We thank the other members of the Galaxy Team (E. Afgan, D. Baker, D.B., D. Bouvier, M. Cech, J. Chilton, D. Clements, N. Coraor, C. Eberhard, J. Goecks, S. Guerler, J. Jackson, G. Von Kuster, R. Lazarus, A.N., J.T.) for their efforts which were instrumental in making this work happen. This project is supported by the NHGRI (HG005542, HG005133, HG004909, and HG006620) and National Science Foundation (DBI 0543285). Additional funding is provided, in part, by a grant from the Pennsylvania Department of Health using Tobacco Settlement Funds. The Department specifically disclaims responsibility for any analyses, interpretations, or conclusions.
- © 2015 Cold Spring Harbor Laboratory Press








