
Mapping Protein–Protein Interactions Using Yeast Two-Hybrid Assays
- Center for the Study of Biological Complexity, Virginia Commonwealth University, Richmond, Virginia 23284
Abstract
Yeast two-hybrid (Y2H) screens are an efficient system for mapping protein–protein interactions and whole interactomes. The screens can be performed using random libraries or collections of defined open reading frames (ORFs) called ORFeomes. This protocol describes both library and array-based Y2H screening, with an emphasis on array-based assays. Array-based Y2H is commonly used to test a number of “prey” proteins for interactions with a single “bait” (target) protein or pool of proteins. The advantage of this approach is the direct identification of interacting protein pairs without further downstream experiments: The identity of the preys is known and does not require further confirmation. In contrast, constructing and screening a random prey library requires identification of individual prey clones and systematic retesting. Retesting is typically performed in an array format.
MATERIALS
Reagents
Adenine (optional, for mating; see Step 41)
Carrier (sheared salmon sperm) DNA (10 mg/mL) (Life Technologies)
-
Store carrier DNA at −20°C. Boil for 10–12 min and cool on ice 3–4 min before use.
Ethanol (≥95% v/v)
Glycerol
Lithium acetate (LiAc) (0.1 m in TE), freshly prepared
-
Immediately before use, combine 1 mL of 1.0 m LiAc with 1 mL of 10× TE buffer (0.1 m Tris–HCl, 10 mm EDTA [pH 7.5]) and 8 mL of dH2O.
PCR and DNA sequencing reagents (for identification of interacting preys in library screens)
PEG (40%)
-
Prepare a 44% PEG solution in advance by dissolving 44.0 g of polyethylene glycol 3000 (PEG) (Sigma-Aldrich) in dH2O for a total volume of 100 mL, and then sterilize by autoclaving. Prepare 40% PEG immediately before use (Step 7) by combining 10 mL of 44% PEG with 1 mL of freshly prepared 0.1 m LiAc (prepared in TE).
Plasmid DNA constructs containing bait protein(s) of interest, plus empty vector control
-
Gateway cloning has become the method of choice for vector assembly; this allows bait constructs (DNA-binding domain [DBD]-ORF fusions) to be generated by transfer of ORFs from entry vectors into specific bait vectors via recombination. We use the bait vectors pGBGT7g (a gentamycin-resistance-encoding version of pGBKT7g) and pGBKCg (Stellberger et al. 2010); see Table 1 and Figure 1. See Landy (1989) and Walhout et al. (2000) for more details regarding Gateway cloning and vectors.
-
Ensure that bait plasmids are compatible with prey plasmids, especially with respect to markers.
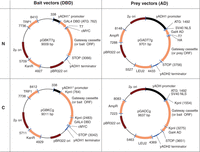
Selected bait and prey vectors. The pGBKT7g and pGADT7g generate amino-terminal DBD and AD fusions, respectively. pGBKCg and pGADCg fuse the DBD or AD at the carboxyl terminus of inserted ORF. (Reprinted from Stellberger et al. 2010.)
Y2H bait and prey vectors and their properties (see also Fig. 1)
Plasmid DNA constructs containing prey proteins of interest (e.g., prey libraries or ORFeomes), plus empty vector control
-
Prey constructs (activation domain [AD]-ORF fusions) must be cloned or obtained in appropriate prey vectors. As with bait vectors, there are several Gateway-compatible prey vectors available, for example, pGADT7g and pGADCg (Stellberger et al. 2010); see Table 1 and Figure 1.
-
Y2H prey libraries for many species, tissues, and cell types are available from several commercial sources or can be constructed in the laboratory using available protocols (Rain et al. 2001). ORFeomes are available for genomes of various sizes, from small viral genomes (e.g., KSHV and VZV [Uetz et al. 2006]) to the bacterial genomes of Escherichia coli, Bacillus anthracis and Yersinia pestis (Rajagopala et al. 2010). Bacterial ORFeomes can be obtained from BEI Resources (https://www.beiresources.org) and academic laboratories. Clone sets from multicellular eukaryotes, such as (Lamesch et al. 2004), human (Rual et al. 2004), and Arabidopsis (Gong et al. 2004) have also been described, though not all genes of interest are yet available in convenient vectors. Cloning and Y2H procedures may be automated using 96-well plates such that entire ORFeomes can be processed in parallel.
Selective liquid medium
-
Follow the recipe for selective solid medium for Y2H, but omit the agar.
Selective solid medium for Y2H (including –L, –T, –LT, and –LTH, with or without 3-amino-1,2,4-triazole [3-AT]), prepared in single-well Omnitray plates
-
3-AT (Sigma-Aldrich) is added to selective medium at varying concentrations (0–100 mm) during the bait self-activation test (Steps 18–23). The 3-AT concentration in the selective (–LTH) medium used for protein interaction screening (Steps 33 and 43) will depend on the results of the self-activation test.
-
Selective solid medium can also be prepared in Qtray plates (Molecular Devices); see Step 15. If plates containing solid medium are wet, allow them to dry for a few hours or overnight at 30°C before plating cells.
Yeast strains (haploid, mating-competent)
-
AH109 (for baits)
-
The AH109 genotype is MATa, trp1-901, leu2-3, 112, ura3-52, his3-200, gal4Δ, gal80Δ, LYS2::GAL1UAS-GAL1TATA-HIS3, GAL2UAS-GAL2TATA-ADE2, URA3::MEL1UAS-MEL1TATA-lacZ (after James et al. 1996).
-
Y187 (for preys)
-
The Y187 genotype is MATα, ura3-52, his3-200, ade2-101, trp1-901, leu2-3, 112, gal4Δ, met−, gal80Δ, URA3::GAL1UAS-GAL1TATA-lacZ (after Harper et al. 1993).
YEPAD liquid medium
-
Follow the recipe for YEPAD solid medium for Y2H, but omit the agar.
YEPAD solid medium for Y2H, prepared in single-well Omnitray plates
-
If plates containing solid medium are wet, allow them to dry for a few hours or overnight at 30°C before plating cells.
Equipment
Aluminum foil
Centrifuge
Centrifuge tubes (15- and 50-mL)
High-density plate replicator (96-pin and/or 384-pin) (Beckman Coulter [Biomek FX 96/384 HDR Tool Body] or V&P Scientific)
-
Solid pins of 0.4–0.5 mm or 1.1–1.6 mm (“thin” or “thick” pins, respectively) for use with the plate replicator are also available from Beckman Coulter or V&P Scientific.
-
Before use and between all transfers, sterilize the plate replicator by dipping the pins into the following series: 20% bleach for 60 sec, sterile H2O for 20 sec, 95% ethanol for 60 sec, and sterile H2O again for 20 sec.
Incubator at 30°C
Incubator/shaker at 30°C, for aerating liquid cultures
-
A benchtop shaker placed inside a standard incubator may also be used.
Laboratory automation robot (Beckman Coulter Biomek 2000, FX or FXP) (optional)
-
A robotic workstation may be used to speed up the screening procedures and maximize reproducibility. Automation is strongly recommended if many clones need to be tested and for screening large numbers of baits, given that manual work is not only boring, straining, and error prone, but also time consuming and tedious. However, maintaining and operating robots can also be expensive and may justify collaboration with a laboratory already in possession of automation equipment.
Microcentrifuge
Microcentrifuge tubes (1.5-mL)
Microtiter plates (96-well, flat-bottom) (Corning)
Omnitray plates (single-well, 86 × 128-mm, with lids) (Nunc)
Parafilm
PCR and DNA sequencing equipment (for identification of interacting preys in library screens)
Spectrophotometer
Vortex mixer
Water bath at 42°C
METHOD
-
A rationale for the selection of library- or array-based Y2H is provided in Introduction: The Yeast Two-Hybrid System: A Tool for Mapping Protein–Protein Interactions (Mehla et al. 2015).
Transforming Yeast
-
This method is suitable for transformation of bait or prey plasmid clones into their respective mating-competent, haploid yeast strains: Prey vectors are transformed into Y187, mating type “α,” while bait vectors are transformed into AH109, mating type “a.” The following is optimized for 100 transformations; a success rate of 100% per transformation should be expected. It also can be scaled down or up as required for high-throughput transformation, and most steps can be automated. As with all high-throughput procedures, small discrepancies can have noticeable downstream consequences. Verify the sterility and pH of reagents and the phenotype of yeast strains before transformation.
-
1. Inoculate 10–20 mL of YEPAD liquid medium with the appropriate haploid yeast strain. Grow overnight at 30°C in a shaking incubator (∼200 rpm, depending on model). After 12–16 h, measure the absorbance (OD600) of the culture.
-
The OD600 should be >1.5.
-
-
2. Dilute the cells in 100 mL of fresh liquid YEPAD medium to an OD600 of ∼0.2–0.3. Grow the cells for ∼3–4 h (depending on the generation time of the strain) at 30°C in a shaking incubator (∼200 rpm).
-
The OD600 should be between 0.5 and 1.0 (at least one doubling), indicating that the cells are in log phase of growth.
-
-
3. Transfer the culture to two 50-mL tubes. Collect the cells by brief centrifugation (∼700g for up to 5 min at room temperature) and discard the supernatant.
-
4. Wash the cells once with 25–50 mL sterile dH2O. Collect the cells by brief centrifugation (∼700g for up to 5 min at room temperature) and discard the supernatant.
-
5. Resuspend the cells in 1 mL of 0.1 m LiAc (prepared in TE) and incubate on ice for 15 min.
-
6. Transfer the suspension to 1.5-mL microcentrifuge tubes and briefly centrifuge (∼350g for 30–60 sec). Discard the supernatant. Resuspend the cells in 1 mL of 0.1 m LiAc (prepared in TE) and incubate them on ice for another 15–20 min.
-
7. Prepare a transformation mix for ∼100 transformation reactions as follows.
-
i. Add 250 µL of carrier DNA (10 mg/mL) to 10 mL of 40% PEG in a sterile 50-mL tube. Vortex for at least 30 sec.
-
ii. Add 1–2 mL of freshly prepared competent yeast cells from Step 6. Vortex for an additional 60 sec.
-
-
8. Transfer 100 µL of reaction mix to each well of a 96-well plate using a multichannel pipette or robotic liquid handler.
-
Depending on the number of samples, the final volume can be divided such that each volume does not exceed 120 µL per well.
-
For fewer samples, 1.5-mL tubes may be used.
-
-
9. Add at least 100 ng of the desired plasmid DNA construct to each well and mix well with the pipette tip. Include one negative control containing carrier DNA but no plasmid DNA and at least one positive control containing the empty vector construct.
-
10. Seal the 96-well plate with adhesive aluminum foil and Parafilm to secure the edges.
-
All exposed plate surfaces should be sealed to protect against contamination.
-
-
11. Vortex the plate gently for 3–4 min, ensuring that the wells do not cross-contaminate.
-
12. Place the plate for 45 min at 30°C with gentle shaking (∼60 rpm).
-
13. Transfer the plate to a water bath at 42°C and incubate for 30 min. Ensure the plate is floating and water is not entering the wells.
-
For small scale transformation using microcentrifuge tubes, this incubation may be reduced to 15 min.
-
-
14. After 30 min, centrifuge the plate at ∼700g for 8–10 min at room temperature. Discard the supernatant by multichannel pipette and rinse the plate surface with 95% ethanol to avoid cross-contamination between wells.
-
15. Resuspend the contents of each well in 50–100 µL of sterile dH2O. Spread each volume on a separate Omnitray plate (or distinct sections of a Qtray) containing selective solid medium.
-
16. Incubate the plates for at least 2–3 d at 30°C.
-
Some strains, such as Y187, may require 3–4 d to yield noticeable transformant growth.
-
Plates may be stored for several months at 4°C as long as the medium does not dry out or become contaminated. For long-term storage, transformants may be cultured in rich (YEPAD) or selective liquid medium overnight, resuspended in 20% glycerol, and transferred to a freezer at −80°C.
-
-
17. After preparation of the desired bait transformants, proceed to self-activation testing of baits (Steps 18–23) followed by library-based (Steps 24–35) and/or array-based (Steps 36–44) screening.
Self-Activation Testing of Bait Transformants Using 3-AT
-
Performing self-activation testing before Y2H screening helps optimize screen conditions, saving time and resources. 3-AT is a competitive inhibitor of the HIS3 gene product and is frequently used in Y2H screening to increase the stringency of selection when using the HIS3 reporter gene. The following assay measures the background activity of the HIS3 reporter in baits in the presence of empty prey vector.
-
18. Prepare 5-mL cultures in YEPAD liquid medium using empty-vector transformants in the prey strain (i.e., the Y187 strain containing the pGADCg empty vector). Grow the cultures for 12–16 h at 30°C in a shaking incubator (∼200 rpm).
-
19. Plate the bait clones on YEPAD using a sterilized plate replicator.
-
Either the standard 96-spot or 384-spot format can be used, depending on the number of clones.
-
-
20. Perform yeast mating between the bait clones and the prey strain carrying empty vector as follows.
-
i. Transfer the empty-vector prey culture from Step 18 to an empty Omnitray plate.
-
ii. Touch the liquid culture with the pins of a sterilized plate replicator, then touch the pins directly onto the plated bait spots from Step 19.
-
iii. Incubate the cells for 36–48 h at 30°C to allow mating.
-
-
21. To select for diploid colonies, transfer the colonies to plates containing the appropriate double-dropout selective medium (–LT) using a sterilized plate replicator. Grow the cells for 2–3 d at 30°C until the colonies are 1 mm in diameter.
-
22. To measure HIS3 reporter gene activity, transfer the diploid colonies to plates containing the appropriate triple-dropout selective medium (–LTH) plus 3-AT at the following concentrations: 0, 1, and 3 mm.
-
The initial 3-AT concentrations may be increased to 10, 25, 50, or 100 mm if necessary. Higher 3-AT concentrations are inadvisable as they will halt all growth.
-
-
23. Incubate the selective plates for ∼1 wk at 30°C. Record the lowest 3-AT concentration at which background colony growth is completely prevented.
-
The recorded concentration of 3-AT should be added to selective plates in the subsequent interaction screens to suppress reporter activation in the absence of an interacting prey.
-
Screening for Protein Interactions Using Genomic/cDNA Libraries/ORFeomes
-
In random library screening (Fig. 2), a single bait of interest is screened against a full library of prey proteins in a single screen. The interacting prey partner is identified by sequencing.

Y2H random library screen. The bait construct (DBD fusion protein) and the prey library (AD fusions with a cDNA or random genomic library) in haploid yeast strains are mixed in a 1:1 ratio. The mating culture is plated on a YEPAD agar plate. After incubation of mating plates for 6 h at 30°C, the diploid cells are transferred to –LTH plates (with or without 3-AT) for selection. The interacting preys are then identified by PCR amplification and sequencing of the vector inserts.
Preparing the Prey and Bait Cultures (Day 1)
-
24. Prepare an activated prey library as follows.
-
i. Inoculate 200 mL of selective liquid medium with an aliquot of the yeast strain containing the prey library at 30°C.
-
ii. Incubate the culture at 30°C overnight with shaking (180 rpm). After ∼16 h (or the next day), measure the absorbance (OD600) of the culture.
-
The OD600 should reach 0.9–1.0 before mating.
-
-
-
25. Prepare a bait liquid culture and negative control as follows.
-
i. Inoculate ∼10 mL of selective liquid medium with the yeast strain containing the bait construct at 30°C.
-
ii. In parallel, start a negative control culture by inoculating identical selective medium with yeast carrying empty prey vector.
-
iii. Incubate overnight at 30°C with shaking (180 rpm).
-
Mating Cells (Day 2)
-
26. Mix the bait and prey construct-containing cultures in equal proportion (1:1 ratio by volume) in 15-mL tubes. For every bait of interest, include a negative control by mating the bait with the empty-vector prey transformant at a 1:1 ratio.
-
27. Pellet the cells by centrifugation at ∼3000g for 2 min. Discard the medium.
-
28. Resuspend the cells in 500 µL of fresh YEPAD liquid medium.
-
29. Plate the cells on YEPAD solid medium. Incubate the plates for 6 h at 30°C or at room temperature overnight.
-
30. Wash the plate with 2 mL of sterile dH2O to collect the cells. Pellet the cells as in Step 27.
-
31. Resuspend the cells in 2 mL of sterile dH2O. Pellet the cells as in Step 27, and resuspend in 2 mL of selective medium (–LTH).
-
32. Check the mating efficiency as follows.
-
i. Prepare a 1:100 dilution in liquid medium and plate the cells on –L, –T, and –LT plates.
-
ii. Incubate the plates for 48–72 h at 30°C. Count the total number of resulting colonies on each type of selective medium.
-
For both diploid (those on –LT) and haploid (those on –L or –T) colonies, viability is the colony count (cfu) per ml (adjusted by the 1:100 dilution factor). Mating efficiency can be calculated by dividing the diploid viability by the lower of the two haploid viability results.
-
Viability of diploids is generally at least 95% that of haploid bait or prey cultures but varies by strain.
-
-
-
33. Plate 100–150 µL of cells on plates containing selective solid medium (–LTH with or without 3-AT).
-
The appropriate concentration of 3-AT in selective medium should be determined using the bait self-activation test (Steps 18–23).
-
-
34. Incubate the plates for 6–8 d at 30°C or until the colonies are ∼1 mm in diameter.
-
35. Select Y2H-positive colonies. Identify the interacting prey partner by amplifying the insert in the prey vector using colony PCR (Rajagopala and Uetz 2011) followed by sequencing.
-
Library screens usually yield dozens of positives that should be retested using array-based Y2H.
-
Screening for Protein Interactions Using Arrays
-
After baits are screened for potential self-activation (Steps 18–23), they may be tested for interactions versus the prey array as follows. Arrays are used as a primary screen or to validate the positives resulting from a library screen. Given the built-in controls present in array-based screens, they are the best choice for differentiating strong and weak interactions from background and efficiently reducing false positives. Usually, preys rather than baits are arrayed in a defined order, as described here, due to the high self-activation potential among baits; the activation domain (AD) used with prey proteins does not generally produce self-activation. An overview of array-based screening is provided in Figure 3.
-
Whether screening is manual or automated, a high-density (96-pin or 384-pin) replicating tool is used for routine transfer of colonies from one plate to another. In the procedure described here, the prey array is gridded on single-well (86 × 128-mm) Omnitray plates in a 96- or 384-colony format.
-
36. After preparation of the desired prey transformants (each carrying one specific AD-containing plasmid), array the preys on plates containing selective solid medium (–L) in either a 96- or 384-format in duplicates or quadruplicates.
-
Baits can be arrayed likewise, as needed. Other array formats such as 768 or 1536 colonies per plate can be used, but the higher densities may render scoring positives more difficult.
-
For long-term storage, all arrays may be cultured in rich (YEPAD) or selective liquid medium overnight, resuspended in 20% glycerol, and transferred to a freezer at −80°C. For day-to-day use, the arrays can be stored on solid selective medium (−L or −T) for up to 3 mo at 4°C. Fresh array plates may be produced using these storage plates before each screen. Copying the array onto fresh selective plates every 2 wk should protect against plasmid loss.
-
-
37. Use the sterile plate replicator to transfer the yeast prey array to plates containing YEPAD solid medium. Grow the array overnight at 30°C.
-
Alternatively, a fresh prey array can be prepared in YEPAD liquid medium in a 96-well plate. Note that only the master prey array (Step 36) should be used to make fresh copies for mating.
-
-
38. Transfer 15–20 mL of liquid YEPAD medium to a 50-mL tube. Inoculate the medium with the bait strain (or pool of baits) and grow overnight at 30°C in a shaking incubator (at ∼200 rpm).
-
A bait pooling strategy may be used to reduce the time of Y2H screening. The pool of baits may be grown together in 15–20 ml YEPAD broth overnight.
-
If the bait strains are frozen, they should be streaked or pinned on selective solid medium plates and grown for 1–2 d at 30°C before any experiment. A fresh bait culture should always be prepared for Y2H mating, as extended culturing in rich media may result in plasmid loss.
-
-
39. Dip the sterile plate replicator in the bait liquid culture and transfer the cells onto a plate containing YEPAD solid medium. Replicate as needed. Allow the spots to dry for 15–20 min.
-
Ensure YEPAD plates are free of residual moisture before pinning to ensure consistent colony formation.
-
-
40. Touch the fresh prey array cultures from Step 37 with the sterilized plate replicator and transfer them directly onto the plated bait spots. Ensure that each of the bait spots receives different prey cells (i.e., a different AD-fusion protein).
-
41. Incubate the cells for 36–48 h at 30°C to allow mating.
-
Supplemental adenine in the bait culture medium may increase the mating efficiency of some baits. Optimization of this step is subject to yeast strains, bait and prey vectors, and laboratory conditions.
-
-
42. To select diploid cells, transfer the colonies from the mating plates to plates containing double-dropout selective solid medium (–LT) using the sterilized plate replicator. Grow the cells for 2–3 d at 30°C until the colonies are 1 mm in diameter.
-
This step ensures successful mating, as only diploid cells containing the selection markers in the prey and bait vectors will grow. Low mating efficiency will noticeably reduce the quality of screen results by obscuring signal and noise.
-
-
43. Transfer the colonies from –LT plates to plates containing triple-dropout selective solid medium (–LTH, with or without 3-AT) using the sterilized plate replicator. Incubate the plates for 6–8 d at 30°C.
-
The choice of 3-AT concentrations will depend on the results of the bait self-activation test (Steps 18–23).
-
-
44. Examine the plates and score the positive interactions by counting colonies that are significantly large in size compared with background. If colonies have been plated in duplicate or quadruplicate, at least half of the colonies in each set should be significantly larger than background growth to indicate a positive result.
-
Plates should be examined and monitored every day for positive colonies and contamination. Most Y2H positive colonies should appear within 3–5 d but some positive interactions may require more time. For most screens, observing >10 positive results per set of 96 different preys constitutes a suspicious result and may indicate false positives.
-
See Troubleshooting.
-

Array-based two-hybrid assays. Bait (pGBKCg, pGBGT7g) and prey (pGADCg, pGADT7g) constructs are transformed into haploid yeast cells of opposite mating type (a and α). Preys are arrayed on selective solid medium (–L). Baits should be tested for self-activation. The Y2H screening begins with mating, in array format, of two haploid strains carrying bait and prey plasmids. Diploid cells are selected by transferring the mated array to double-dropout (–LT) followed by triple-dropout (–LTH) plates. The rectangles on the selective plate (bottom panel) mark negative and positive interactions between the bait and the prey at these specific positions of the array.
TROUBLESHOOTING
Problem (Step 44): There are high background growth levels or false positives due to self-activation of baits.
Solution: Some baits can activate transcription of the reporter gene (e.g., HIS3) in the absence of any prey partner. Bait self-activation of the HIS3 reporter gene can be suppressed by adding 3-AT to the selective medium. A concentration of 1–10 mm is usually sufficient to alleviate most self-activation, though up to 100 mm may be necessary in some cases.
Cloning the bait gene into a different vector can also reduce self-activation. Self-activating baits expressed as amino-terminal fusion products (i.e., in pGBGT7g) may show no self-activation at all when expressed as carboxy-terminal fusions (i.e., in pGBKCg). Alternatively, a bait construct can be modified to remove the region responsible for the self-activation. This step may be necessary for baits which strongly activate transcription.
Problem (Step 44): There are high background growth levels or false positives due to nonreproducible, seemingly random sets of positive results.
Solution: The main limiting factor associated with the Y2H assay in general is the potential for a large number of false positives. Without proper controls, >90% of all interactions in a Y2H screen can be the result of nonreproducible background (Uetz 2002). The following can be used to increase reliability and confidence in Y2H interaction data sets.
-
Retesting all positive interactions by repeated mating usually reduces the number of false positives and provides confirmation of positive interactions. Interactions may be repeated in quadruplicate or by mating the interacting pair and scoring its signal strength with reference to empty prey vector.
-
The use of different vector combinations can provide additional evidence for positive interactions in multiple screens, especially when protein interactions are expected to be inhibited by certain conditions (i.e., when a binding domain is only exposed in a particular binding conformation). This protocol was designed for use with combinations of the vectors used by Stellberger et al. (2010) (Fig. 1).
-
Potential false-positives also can be retested by employing a full set of amino-terminal and carboxy-terminal protein fusions (Stellberger et al. 2010).
-
Alternative methods, such as β-galactosidase assays, are also available for further confirmation of Y2H interactions; see the protocols by Serebriiskii et al. (2005) and Rajagopala et al. (2007).
- © 2015 Cold Spring Harbor Laboratory Press








