
Calling Card Analysis in Budding Yeast
- Department of Genetics, Center for Genome Sciences and Systems Biology, Washington University School of Medicine, St. Louis, Missouri 63110
Abstract
Calling card analysis is a high-throughput method for identifying the genomic binding sites of multiple transcription factors in a single experiment in budding yeast. By tagging a DNA-binding protein with a targeting domain that directs the insertion of the Ty5 retrotransposon, the genomic binding sites for that transcription factor are marked. The transposition locations are then identified en masse by Illumina sequencing. The calling card protocol allows for simultaneous analysis of multiple transcription factors. By cloning barcodes into the Ty5 transposon, it is possible to pair a unique barcode with every transcription factor in the experiment. The method presented here uses expression of transcription factors from their native loci; however, it can also be altered to measure binding sites of transcription factors overexpressed from a plasmid.
MATERIALS
Reagents
Agarose gels (0.7%) and running buffer
Barcoded Ty5 plasmid (pRM1001-series; 200 ng/μL)
-
These plasmids, which are available from the corresponding author, each contain an 8-bp barcode within the transposon that will be matched to each tagged strain.
Betaine (5 m)
BSA (20 mg/mL; molecular biology grade)
Carrier (Sheared Salmon Sperm) DNA Solution (10 mg/mL; Invitrogen)
Chloroform
dH2O
dNTP mix (10 mm each of dATP, dCTP, dGTP, dTTP)
Ethanol (70%, 100%)
Genomic DNA from yeast strain yRM1009
-
This strain is available from the corresponding author.
Glucose − His 5-fluoroorotic acid (5-FOA) agar plates
Glycerol (20%; optional; see Step 16)
HindPI1 restriction enzyme and appropriate buffer
HpaII restriction enzyme and appropriate buffer
Lithium acetate (1 m)
Phenol:choloroform:isoamyl alcohol (PCA) (25:24:1)
Phusion DNA polymerase
Phusion HF buffer (5×)
Polyethylene glycol 3500 (PEG 3500; 50% w/v)
Primers (all sequences are listed 5′ to 3′)
-
“Forward” cloning primer: (N)40AGAGTGTCGCATAGTGATAC
-
This primer includes 40 bp upstream of the stop codon of the ORF of the DNA-binding protein of interest (without including the stop codon) plus 20 bp to amplify the tagging domain.
-
“Reverse” cloning primer: (N)40CGCACTTAACTTCGCATCTG
-
This primer includes the reverse complement of the 40 bp downstream from the ORF of the DNA-binding protein of interest (without including the stop codon) plus 20 bp to amplify the tagging domain.
-
“Forward” inverse PCR primer: AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTAATTCACTACGTCAACA
-
“Reverse” inverse PCR primer: CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATC
RedTaq DNA polymerase and associated buffer
RNaseA (20 mg/mL)
Sodium acetate (3 m, pH 5.2)
TaqI restriction enzyme and appropriate buffer
T4 DNA ligase and associated buffer
Yeast calling card lysis buffer
Yeast strain of desired background
-
Strain must be deficient for SIR4, HIS3, and URA3 and sensitive to nourseothricin (NAT) antibiotic.
YPD (liquid medium and agar plates)
-
YPD agar plates with and without 100 µg/mL NAT are needed.
Equipment
Agarose gel apparatus
Centrifuge
Conical centrifuge tubes (15-mL)
Glass beads (0.2 mm diameter)
Glass culture tubes (5-mL; disposable)
Glass yeast spreader
Heat blocks
Incubator (30°C)
Microcentrifuge
Microcentrifuge tubes (1.5-mL)
Microcentrifuge tube rotator
PCR (polymerase chain reaction) tubes
QIAquick PCR purification kit (Qiagen)
Sequencing machine (Illumina GA, Illumina Miseq, or Illumina HiSeq with paired-end capabilities)
Spectrophotometer for DNA quantification (Nanodrop [Thermo Scientific] or Qubit fluorometer [Invitrogen])
Thermocycler
Vacuum concentrator (e.g., Speedvac)
Velvet replica plating pads and base
Vortexer
Water bath
METHOD
Cloning Strain
-
This stage involves cloning the Ty5 integrase-interacting domain of Sir4 to the carboxyl terminus of the desired TF. If the desired tagged strains have already been created, proceed to Step 11. See Figure 1 for an overview of the entire protocol.
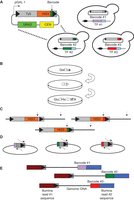
Overview of the calling card protocol. (A) Barcoded Ty5 transposons are separately transformed into yeast strains with TFs tagged with Sir4. (B) The strains are pooled, expression of Ty5 is induced, and the subsequent growth medium selects for genomic insertions. (C) Genomic DNA is digested with restriction endonucleases. (D) Digested fragments are self-ligated. (E) Inverse PCR adds Illumina sequencing adaptors.
-
1. Amplify the Sir4-NatR sequence from genomic DNA of strain yRM1009. Add the following PCR mix components and mix well:
-
1× Phusion HF buffer
-
0.5 µm “forward” cloning PCR primer
-
0.5 µm “reverse” cloning PCR primer
-
0.2 mm of each dNTP
-
1 unit Phusion DNA Polymerase
-
100 ng genomic DNA of yRM1009
-
dH2O to 50 µL
-
-
2. Perform the following cycling program:
No. of cycles Temperature Time 1 98°C 30 sec 30 98°C 10 sec 58°C 30 sec 72°C 2 min 1 72°C 5 min 1 12°C Hold -
3. Run 5 µL of the PCR product on 0.7% agarose gel using standard techniques to confirm success of PCR.
-
A single product at 1.8 kb is expected.
-
-
4. Inoculate the yeast strain of the desired background into 5 mL of YPD in a disposable 5-mL glass culture tube. Grow to saturation overnight at 30°C with shaking. Dilute 50 µL of the saturated starter culture into 5 mL YPD in a disposable 5-mL glass culture tube. Grow at 30°C with shaking to an OD600 between 0.8 and 1.0 (∼ 6 h).
-
5. Pellet the cells by centrifuging the culture tube at 3000 rpm for 2 min at room temperature. Remove the supernatant. Resuspend the cells in 1000 µL of dH2O and transfer the cells to a microcentrifuge tube. Pellet the cells by centrifuging the tube at 10,000 rpm for 2 min at room temperature. Remove the supernatant.
-
6. Add the following components of the transformation mix with the remaining PCR product from Step 3 directly on top of the pelleted cells to the following final concentrations and to a total volume of 360 µL:
Component Amount (per reaction) Final concentration PEG 3500 (50% w/v) 240 µL 33.3% Lithium acetate (1 m) 36 µL 0.1 m Carrier DNA solution 10 µL 250 ng/μL PCR product plus dH2O 74 µL -
7. Vortex the mixture and incubate in a water bath for 40 min at 42°C.
-
8. Centrifuge the tube at 10,000 rpm for 2 min at room temperature and remove supernatant. Resuspend the cell pellet in 1 mL of YPD and incubate with rotation for 60 min at 30°C.
-
9. Centrifuge the tube at 10,000 rpm for 2 min at room temperature and remove YPD. Resuspend the cell pellet in 50 µL dH2O and plate on a YPD agar plate containing 100 µg/mL NAT and incubate for 2 d at 30°C.
-
10. Select NAT resistant colonies and Sanger sequence the DNA bordering the tagged ORF to verify a correct clone.
-
Select at least three clones for each cloned strain.
-
Plasmid Transformation and Induction
-
This stage requires four different growth steps on separate days with 1 h of hands-on time each day.
-
11. Select a unique barcoded Ty5 donor plasmid for each transcription factor–tagged strain to be examined.
-
12. Separately inoculate each tagged strain from step 10 into 5 mL of YPD in a disposable 5-mL glass culture tube. Grow to saturation overnight at 30°C with shaking. Dilute 50 µL of the saturated starter culture into 5 mL YPD in a disposable 5-mL glass culture tube. Grow at 30°C with shaking to an OD600 between 0.8 and 1.0 (∼ 6 h).
-
13. Pellet the cells by centrifuging the culture tube at 3000 rpm for 2 min at room temperature. Remove the supernatant and wash cells as in Step 5.
-
14. For each tagged strain, add the following components of the transformation mix with a barcoded Ty5 plasmid to the following final concentrations and to a total volume of 360 µL:
Component Amount (per reaction) Final concentration PEG 3500 (50% w/v) 240 µL 33.3% Lithium acetate (1.0 m) 36 µL 0.1 m Carrier (Sheared Sperm DNA Solution) 10 µL 250 ng/μL Ty5 plasmid (200 ng/μL) 1 µL 0.6 ng/μL dH2O 73 µL -
15. Vortex the mixture and incubate in a water bath for 40 min at 42°C.
-
16. Centrifuge tube at 10,000 rpm for 2 min at room temperature and remove the transformation mix. Resuspend the cell pellet in 50 µL of dH2O and plate on a Glucose − His plate. Incubate at 30°C for 2 d.
-
Transformed yeast can be frozen in 20% glycerol at −80°C at this step to avoid repeating the previous steps in future experiments.
-
-
17. Inoculate a single colony from each barcoded-plasmid transformed strain in 5 mL of liquid Glucose − Ura medium in a disposable 5-mL glass culture tube. Grow to saturation overnight at 30°C with shaking. Pellet cells by centrifuging the culture tube at 3000 rpm at room temperature for 2 min. Remove medium.
-
18. Resuspend the cells in each tube in 1 mL dH2O. Combine all transformed strains into a single tube and mix well. Plate 100 µL of this mixture onto each Galactose − Ura agar plate and incubate at room temperature for 2 d.
-
Yeast should form a confluent lawn.
-
The Ty5 transposon is induced by galactose, so this is the step at which transcription factor binding is measured. If additional growth conditions are desired in the experiment, these should be supplied at this step. The number of Galactose − Uracil plates should scale with the number transcription factors being studied. As a rule of thumb, use two 10-cm plates for each strain in the experiment (enough for 5 × 103 insertions), but DNA-binding proteins with a very large number of targets may require more plates to gather enough insertions to map all of their binding sites.
-
-
19. Replica plate cells to YPD agar plates by pressing the yeast-side of the Galactose − Ura agar plate to a clean velvet, removing, and then pressing a new YPD agar plate against the velvet. Use a clean velvet for each pair of plates. Incubate the plates for 1 d at 30°C.
-
Yeast should form a confluent lawn.
-
-
20. Replica plate cells from YPD plates to Glucose − His 5-FOA agar plates and incubate for 2 d at 30°C.
-
This step selects for genomic insertions of Ty5 and counter-selects against cells still containing the plasmid. Each 10-cm plate should have ∼2 × 103 colonies by the second day of growth.
-
See Troubleshooting.
-
DNA Extraction
-
Requires a total of 2 h.
-
21. Pipette 1 mL of liquid YPD onto each Glu – His 5-FOA plate and scrape the yeast with a glass yeast spreader to put the cells into solution. Pipette the solution into a 15-mL conical tube. Combine cells from different plates into a single conical tube. Mix by gently inverting the tube.
-
22. Transfer 500 µL of the liquid mixture to a 1.5-mL microcentrifuge tube. Centrifuge at 13,000 rpm for 10 min at room temperature in a microcentrifuge and pour off the liquid.
-
23. To the pelleted cells add:
-
400 µL Yeast calling card lysis buffer
-
400 µL Glass beads (measure using a microcentrifuge tube)
-
400 µL Phenol:choloroform:isoamyl alcohol (25:24:1)
-
-
24. Vortex for 10 min at room temperature.
-
25. Centrifuge at 13,000 rpm for 10 min at room temperature.
-
26. Transfer the aqueous supernatant to a new microcentrifuge tube.
-
27. Add 400 µL of phenol:choloroform:isoamyl alcohol (25:24:1). Repeat Steps 24–26.
-
28. Add 400 µL of chloroform. Repeat Steps 24–26.
-
29. Precipitate DNA by adding 40 µL of 3 m sodium acetate and 1000 µL of ethanol (100%) at room temperature. Vortex and incubate for 60 min at −70°C.
-
30. Centrifuge at 13,000 rpm for 10 min at room temperature. Carefully remove the supernatant.
-
31. Carefully wash the pellet with 500 µL of ethanol (70%) at room temperature.
-
32. Centrifuge at 13,000 rpm for 1 min at room temperature.
-
33. Remove ethanol supernatant with a micropipette and dry the pellet in a vacuum concentrator until all liquid is removed (∼ 10 min). Reconstitute the pellet in 50 µL dH2O and 2 µL RNaseA and incubate for 30 min at 37°C.
-
At this step, genomic DNA can be frozen at −20°C and stored long-term before subsequent steps.
-
Genomic Digestions
-
Requires a total of 3.5 h.
-
34. Measure DNA concentration with a spectrophotometer.
-
35. Put 4 µg of DNA into each of three PCR tubes. Perform the following three digestions in a total volume of 50 µL each with the final concentrations and conditions as listed:
-
i. TaqI digestion
-
4 Units TaqI enzyme
-
1× TaqI-compliant buffer
-
1× BSA
-
dH2O to 50 µL
-
Incubate for 3 h at 65°C followed by a heat inactivation for 20 min at 80°C.
-
-
ii. HindPI1 digestion
-
4 Units HindPI1 enzyme
-
1× HindPI1-compliant buffer
-
dH2O to 50 µL
-
Incubate for 3 h at 37°C followed by a heat inactivation for 20 min at 65°C.
-
-
iii. HpaII digestion
-
4 units HpaII enzyme
-
1× HpaII-compliant buffer
-
dH2O to 50 µL
-
Incubate for 3 h at 37°C followed by a heat inactivation for 20 min at 65°C.
-
-
-
36. Purify digestions using the QIAquick PCR purification kit, following the manufacturer’s protocol. Elute DNA in 50 µL of dH2O.
Self-Ligation
-
Requires a total of 14 h or overnight.
-
37. To each of the eluates from Step 36 add the reagents below to the following final concentrations:
-
18 units T4 ligase
-
1× T4 ligation buffer
-
dH2O to 400 µL
-
-
38. Incubate for at least 12 h at 14°C.
-
Overnight incubation is preferable.
-
-
39. Precipitate DNA by following Steps 29–32.
-
40. Remove ethanol and dry the pellet in a vacuum concentrator. Resuspend the pellet in 50 µL dH2O and vortex.
Inverse PCR
-
Requires a total of 4 h.
-
41. Add the following PCR mix components to half (25 µL) of each of the reconstituted DNA samples from Step 40 to yield the indicated concentrations:
-
1× RedTaq PCR Buffer
-
0.5 µm “forward” inverse PCR primer
-
0.5 µm “reverse” inverse PCR primer
-
0.2 mm of each dNTP
-
0.5 m betaine
-
4 units RedTaq Enzyme
-
dH2O to 50 µL
-
-
42. Perform the following cycling program:
No. of cycles Temperature Time 1 93°C 2 min 30 93°C 30 sec 60°C 6 min 1 12°C Hold -
43. Run 5 µL of each PCR product on a 0.7% agarose gel using standard techniques to confirm the success of PCR.
-
Expect products ranging from 200 bp to 2 kb.
-
See Troubleshooting.
-
-
44. Purify PCR products using QIAquick PCR purification kit, following the manufacturers's protocol. Elute DNA in 50 µL of dH2O and quantify the DNA concentration using Nanodrop/Qubit.
-
45. Combine equimolar quantities of the 3 PCR products and dilute to 10 nm in dH2O for Illumina sequencing.
-
Assume an average PCR product size of 200 bp, which makes 10 nm equal to 1.32 ng/μL. At least 20 µL of the final solution is needed for Illumina sequencing.
-
-
46. Sequence the DNA on a paired-end Illumina sequencing run according to the manufacturer’s instructions.
-
47. Analyze data by demultiplexing reads using the 8-bp barcode (the first eight bases on the second read), removing the first 17 bp of the read (corresponds to the Ty5 transposon), and aligning the remaining bases of the read to the reference genome of the background yeast strain.
-
See Troubleshooting.
-
TROUBLESHOOTING
Problem (Step 20 or 47): A high background of yeast on the Glucose − Histidine + 5-FOA selection step or a high percentage of plasmid reads in the sequencing is observed.
Solution: The 5-FOA selection step is not killing all of the yeast still containing the Ty5 plasmid. An additional round of replica plating on Glucose − Histidine + 5-FOA plates will reduce plasmid in the extracted DNA.
Problem (Step 43): A thick band is observed at 100 bp with a smear of genomic DNA above this product.
Solution: Primer–dimers have formed during the inverse PCR. Perform a QIAquick gel extraction (QIAGEN) to separate the larger products from the primer–dimer size fragments before sequencing.
DISCUSSION
Chromatin immunoprecipitation read out by either microarrays (ChIP-chip) (Horak and Snyder 2002) or next-generation sequencing (ChIP-seq) (Johnson et al. 2007) has been an invaluable technique for identifying the binding sites of transcription factors (TFs) in yeast. For studies requiring large numbers of TFs, the nature of ChIP-based methods requires increased time and work for each additional TF in the experiment. Large sequencing capacities of next-generation sequencers allow for ChIP-seq libraries of many TFs to be barcoded and sequenced in parallel (Lefrançois et al. 2009); however, the immunoprecipitation for each TF is performed independently. This limitation makes large-scale ChIP experiments laborious. The ability of the calling card technique to multiplex many TFs through most of the steps in this protocol makes scaling up these experiments more feasible.
The number of TFs that can be included in this protocol is a function of the downstream sequencing capacity. To ensure adequate coverage of all of the insertions with each barcode, 1−2 × 106 reads are required for each barcode/TF included in the multiplex. Experiments with large numbers of TFs may make the growth and selection steps on solid plates unsuitable. For these larger experiments the protocol can be altered to move these steps (18–20) to liquid cultures. For more information on the data analysis or altering the protocol to overexpress the TF-Sir4 fusion from a plasmid, please see Wang et al. (2011).
A limitation of calling cards is that there are currently fewer existing yeast strains with TFs tagged for the method compared with ChIP-ready tagged strains. The study of some TFs or background strains will necessitate cloning them as described in the first steps of this protocol. The method is easy to use and provides high signal-to-noise mapping of DNA-binding with an accuracy and resolution comparable with ChIP-seq (Wang et al. 2011). This protocol should be adaptable to many experiments for mapping binding sites of DNA-binding proteins, especially when multiple proteins need to be studied.
- © 2016 Cold Spring Harbor Laboratory Press








