
Cell Cycle Synchronization of Schizosaccharomyces pombe by Centrifugal Elutriation of Small Cells
- 1CRUK Cell Division Group, Cancer Research UK Manchester Institute, University of Manchester, Manchester M20 4BX, United Kingdom;
- 2EPFL SV ISREC UPSIM, SV2.1830, CH-1015 Lausanne, Switzerland
Abstract
Division of Schizosaccharomyces pombe by medial fission produces identically sized daughter cells that grow by tip extension until their own division is prompted by reaching the same critical size for division as the parental cell. The fidelity of this size control in the absence of perturbation means that cells of the same size are at the same point in the cell cycle. Size selection of small cells from an asynchronous culture by centrifugal elutriation permits generation of synchronous cultures large enough for biochemical analysis. The changes observed in the synchronized cell cycle progression of such cultures are representative of those that accompany cell cycle progression of individual cells. Here, we describe how size selection with the Beckman Coulter JE-5.0 rotor can be used to generate synchronized cultures. Because of the continuous passage of medium through the rotor throughout the procedure, elutriation is considered to have less impact on the integrity of the cell cycle than other approaches. Two protocols are presented here: The first generates a 2-L culture ideal for detailed biochemical analysis, whereas the second allows rapid generation and simultaneous analysis of three smaller (200-mL) cultures.
MATERIALS
Reagents
Calcofluor-white (saturated solution in phosphate-buffered saline) for staining as described in Protocol: Chromatin and Cell Wall Staining of Schizosaccharomyces pombe (Hagan 2016)
EMM2 (liquid medium, with appropriate supplements)
-
Prepare 7 L for the initial culture plus 3 L (prewarmed to culture temperature) for elutriation.
Formalin solution (30%)
Schizosaccharomyces pombe cells in growth medium (400-mL preculture at mid-log phase [5 × 106 cells/mL])
-
When growing mutant strains in minimal medium (including tagged versions of otherwise wild-type genes), use prototrophs wherever possible.
Equipment
Bottles (1-L)
Centrifuge (modified Beckman Coulter J6- or Avanti J-26 series)
Centrifuge tubes (50-mL, screw-cap)
Drip tray and 500-mL bottle for exhaust tube setup
Elutriator rotor (Beckman Coulter JE-5.0) and anchor cable
-
Two chambers can be used with the Beckman Coulter JE-5.0 elutriator rotor: The 40-mL chamber generates cultures between 1 and 3.2 L, whereas the 4-mL chamber can generate three 200-mL cultures in 1 h; see Method.
-
Clean the rotor with 2 L of H2O after the procedure.
Epi-fluorescence microscope with filter for imaging calcofluor (excitation 450 nm, emission 347 nm)
Flasks (5-L)
Microcentrifuge tubes (1.5-mL)
Microscope slides and coverslips
Phase contrast microscope with 40× objective (as needed; see Step 14)
Pump (e.g., Cole Palmer 900-292)
-
Calibrate the pump before use by establishing settings to match the pump rates provided in the Method.
Retort stand with clamps to hold bubble trap and pressure gauge
-
The bubble trap is called the “sample reservoir” in the manual from Beckman Coulter.
Shaking air incubator, with four clamps for 5-L flasks fitted to platform, at culture temperature
Shaking water bath, with any clamps removed from platform, at culture temperature
Spectrophotometer and cuvettes (optional; see Step 11)
METHOD
Using the 40-mL Chamber for Elutriation
-
For the following procedure, 2.7 × 1010 cells are required to yield 1.6–2.5 L of cells at 1 × 106 cells/mL.
Cell Preparation and Setup
-
1. At least 20 h before elutriation, dilute the 400-mL preculture (5 × 106 cells/mL) in EMM2 in 5-L flasks to produce 7 L of culture at 3.8 × 106 cells/mL on the day of elutriation. Do not exceed 1.8 L per flask. Grow the culture with rotation at 240 rpm or faster for at least 20 h (until Step 7).
-
Culture conditions are critical. If cell density exceeds 3.8 × 106 cells/mL, synchrony will be compromised in the second cycle; this suggests that cells from a higher culture density are more prone to starvation in the separation chamber. If density is far below 3.8 × 106 cells/mL, the reduction in the robustness of the gradient will compromise size selection. Similar issues apply to culture volume, speed, and late inoculation.
-
-
2. Secure the assembled rotor in the centrifuge with an anchor cable, clamp the culture supply and efflux tubes to the cable, and check that the tubes will remain attached to the rotor when the anchor cable is taut. Assemble the other components according to Figure 1A. Set the centrifuge temperature to match the culture temperature.
-
If elution is to be performed at 36°C, store the assembled rotor overnight in an incubator at 36°C.
-
-
3. Label 31 microcentrifuge tubes from 0 to 500 in increments of 20. Aliquot 50 µL of 30% formalin into each tube before closing the lids.
-
4. Label 50-mL centrifuge tubes for sample collection in Step 11.
-
Cell mass increases exponentially; hence, the volume required to maintain biomass will decrease exponentially, and so the volume sampled at each point throughout the culture should decrease exponentially (i.e., if 50 mL is collected at t = 0 at 25°C, then 37.5, 21, 15.75, and 11.8 mL will produce the same yield at each 100-min interval).
-
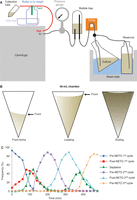
(A) The configuration of the elutriation system. Input into the pump can be switched between a reservoir bottle containing medium and the 5-L flask containing culture. The bubble trap is called the “sample reservoir” in the manual from Beckman Coulter. The exhaust tube from the rotor (red) is taped to the top of an empty bottle inside a drip tray during elution to ensure the height of the tube remains fixed, as the simple act of raising or lowering the height of the tube will move the front back and forth on the rotor. (B) Depictions of the appearance of the cells in the 40-mL chamber at the indicated stages of the protocol. (C) Frequency of septation and pre- and post-NETO cells as size-selected cells transit two consecutive cell cycles. NETO is delayed in the G2 population isolated by size selection.
Elutriation
-
5. On the day of elutriation, place a bottle of fresh EMM2 in the water bath at the culture temperature. Pump prewarmed EMM2 through the system at 160 mL/min with the exhaust tube from the rotor returning to the same bottle. Remove the bubble trap from its clamp and invert so the inlet is uppermost until the trap is 60% full of medium, and then reinvert and clamp with inlet below (leaving a large air pocket in the bubble trap).
-
6. To purge the rotor of bubbles, set the rotor speed to 2000 rpm and press Start. If the “back pressure” reaches 8 psi, press stop immediately and check the connections. Stop the centrifuge as soon as the speed reaches 2000 rpm.
-
Small bubbles will emerge when the speed returns to 130–100 rpm.
-
-
7. Begin cell loading as follows.
-
i. Set the rotor speed to 4000 rpm while maintaining the pump rate at 160 mL/min. At 4000 rpm, adjust the stroboscope to see the chamber.
-
ii. Place a culture flask from Step 1 next to the bottle of medium in the water bath.
-
iii. If eluting at 36°C, run prewarmed medium through the rotor for 20 min before loading cells.
-
iv. Switch the input from fresh medium to the culture flask. Move the tube for the exhaust medium from the rotor to a waste vessel.
-
The chamber will become increasingly cloudy until enough cells accumulate to condense into a dense mass in the lower half of the chamber. A clear “front” will separate the mass of cells from the clearer void above it (Fig. 1B, left).
-
-
-
8. Once the front has formed, increase the pump rate to 196 mL/min.
-
The front will move toward the top of the chamber.
-
-
9. Continue loading as follows.
-
i. Periodically check the position of the front and the back pressure. Maintain a small gap between the front and the top of the chamber by slowing the pump rate. (Adjust the strobe to maintain sight of the chamber.) Reduce the pump rate if the back pressure exceeds 5 psi.
-
The front will continuously move toward the top of the chamber as shown in Figure 1B, center.
-
Wild-type cells can tolerate back pressures up to 8 psi; however, the synchrony of mutants can be compromised above 5 psi.
-
See Troubleshooting.
-
-
ii. Periodically replenish the culture from the additional culture flasks and aerate by shaking the feeder culture. If the air pocket in the bubble trap shrinks below 10%, introduce more air by repeatedly lifting the intake tube from the culture.
-
The air pocket in the bubble trap acts as a shock absorber to dampen surges created by the pump.
-
-
-
10. When the medium exiting the rotor is clear (usually 20 min after starting the run), collect 200 mL of this conditioned medium in a bottle and place it in the water bath.
-
11. Twenty-seven minutes after initiating loading, begin elution as follows.
-
After 27 min, the front between the void space and the dark mass of cells will be very stable. If cells spend >27 min in the chamber, the synchrony in the second cycle will be compromised, suggesting that cells begin to starve after 27 min.
-
i. Switch the input from the culture to a shaken bottle of fresh medium and decrease the centrifuge speed by 50 rpm increments until the profile matches that in Figure 1B, right. Tape the exhaust tube across the top of a 500-mL bottle (to keep its height constant throughout elution as in Fig. 1A).
-
The level of synchrony will drop if the height of the collecting tube is not fixed throughout the sampling.
-
-
ii. Begin collecting 50-mL samples in 50-mL centrifuge tubes. Check the cells in each sample by microscopy.
-
Elution of cells can be detected with a spectrophotometer at OD595 = 0.05–0.06; however, microscopy has the advantage of immediate indication of size selection.
-
-
iii. If cells have not begun to elute after 50 mL have been collected, reduce the centrifuge speed by further increments of 50 rpm until they appear.
-
The void volume in the system is 50 mL or slightly more, so always collect 50 mL before making the next change.
-
-
-
12. Once cells are eluting, add 150 mL of the conditioned medium from Step 10 to the reservoir of fresh medium. Continue to collect samples in 50-mL tubes throughout elution, checking the size selection and transferring all good samples to a 5-L flask. Because the physical properties of the gradient change as cells leave, periodically reduce the centrifuge speed by 50-rpm increments to maintain elution. Continue replenishing the reservoir with fresh medium.
-
Addition of conditioned medium guards against the possibility that S. pombe will use small molecules for intercellular communication.
-
-
13. Once the sampling is complete, count the cell number and dilute with medium to 1 × 106 cells/mL. Return the cells to the shaking incubator. For >1.8 L, use two flasks with an identical volume in each. Do not grow >1.8 L of eluted culture in a 5-L flask.
-
The ratio of the culture volume to flask volume sets the precise periodicity of the cell cycle of the size-selected culture.
-
-
14. Every 20 min, remove 500 µL from the culture and transfer it into the appropriately labeled 1.5-mL tube containing formalin for calcofluor staining to monitor septation index as described in Protocol: Chromatin and Cell Wall Staining of Schizosaccharomyces pombe (Hagan 2016). (If using two flasks, take consecutive samples from each flask.)
-
Remember to accommodate the exponential increase in biomass (see Step 4) when taking specific samples.
-
Septation index can also be monitored using a phase contrast microscope by creating a “pseudodark field” illumination. With a 40× phase 2 objective, the phase ring should be moved on the condenser from the phase 2 toward the phase 3 setting until the field of view becomes black, yet the cell outlines and septa shine brightly.
-
See Troubleshooting.
-
Using the 4-mL Chamber for Elutriation
-
For the following procedure, 3.8 × 109 cells are required to yield 150–200 mL of cells at 1 × 106 cells/mL.
-
15. To use the 4-mL chamber for elutriation, follow the procedure for the 40-mL rotor with the following deviations.
-
In Step 5, pump prewarmed EMM2 at 54 mL/min. Raise the speed to 2000 rpm before stopping to purge rotor of bubbles as in Step 6.
-
In Step 7, set the rotor speed to 4700 rpm and increase the pump rate to 196 mL/min to fill the bubble trap with culture. Reduce to 50 mL/min immediately afterward. Load the cells at 50 mL/min until a front forms (~2 min).
-
In Step 8, once the front has formed, increase the pump rate to 78 mL/min.
-
In Step 9, keep the front away from the very top of the chamber during loading by reducing the pump rate back toward 50 mL/min.
-
In Step 10, 10 min after initiating loading, collect 50 mL of conditioned medium in a bottle.
-
In Step 11, 15 min after initiating loading, switch the input from the culture to fresh medium and elute the cells by increasing the pump rate by 4.5 mL/min. If cells do not elute, increase in further 4.5-mL/min increments until they do. Once cells are eluting, increase by increments of 2.25-mL/min to maintain elution.
-
Unlike the profile using the 40-mL chamber, the front will no longer be visible when cells are eluting.
-
-
TROUBLESHOOTING
Problem (Step 9.i): Cells clump in the chamber, plug the inlet, and cause a catastrophic rise in back pressure that forces the input tube from the rotor.
Solution: This can be addressed with the following modifications.
-
Outcross any auxotrophic markers and repeat the procedure.
-
Use smaller culture volumes in the main and preculture flasks to increase aeration.
-
Change the settings to reduce the stress (i.e., use centrifuge speeds below 3600 rpm with compensatory changes in the pump rate).
-
If the problem persists and your experiment remains meaningful in rich medium, switch to yeast extract medium.
Problem (Step 14): The synchrony of the first cell cycle is okay (with a sharp peak of septating cells), but the second is poor (with a broad septation peak).
Solution: This is commonly seen when either the preculture or the starter culture exceeded mid log-phase, or the culture was inoculated too late the preceding day. Repeat the experiment paying more attention to the culture conditions, or try the following.
-
Try the solutions detailed above for Troubleshooting: Step 9.i.
-
Try eluting 23 min after culture loading is initiated.
-
Check new end take off (NETO) timing with calcofluor staining (Mitchison and Nurse 1985). Randomization of NETO makes growth rates too heterogeneous for synchronization.
Problem (Step 14): No cell division occurs after elution.
Solution: Mutation confers acute sensitivity to elutriation stress. Try the solutions detailed above for Troubleshooting: Step 9.i, and/or try a different-sized chamber, as stresses may differ between the two rotors.
DISCUSSION
Elutriation is considered the least perturbing of any synchronization method, because cells are constantly bathed in fresh, aerated medium; however, any manipulation perturbs the cell cycle to some degree. The centrifugal forces of elutriation mildly activate both the Sty1 stress and the Chk1 DNA integrity pathways and delay NETO in the first division after arrest (Fig. 1C). Consequently, it is important to study the cell cycle controls in the second cycle and not the first after elutriation.
ACKNOWLEDGMENTS
The V.S. Laboratory is funded by Ecole Polytechnique Fédérale de Lausanne (EPFL) and Swiss National Science Foundation. I.M.H. and A.G. are funded by Cancer Research UK (CRUK) C147/A6058.
Footnotes
-
↵3 Correspondence: Iain.Hagan{at}cruk.manchester.ac.uk
-
From the Fission Yeast collection, edited by Iain M. Hagan, Antony M. Carr, Agnes Grallert, and Paul Nurse.
- © 2016 Cold Spring Harbor Laboratory Press








