
Characterizing Antibodies
Abstract
Perhaps because they are such commonly used tools, many researchers view antibodies one-dimensionally: Antibody Y binds antigen X. Although few techniques require a comprehensive understanding of any particular antibody’s characteristics, well-executed experiments do require a basic appreciation of what is known and, equally as important, what is not known about the antibody being used. Ignorance of the relevant antibody characteristics critical for a particular assay can easily lead to loss of precious resources (time, money, and limiting amounts of sample) and, in worst-case scenarios, erroneous conclusions. Here, we describe various antibody characteristics to provide a more well-rounded perspective of these critical reagents. With this information, it will be easier to make informed decisions on how best to choose and use the available antibodies, as well as knowing when it is essential and how to determine a particular as yet-undefined characteristic.
ANTIGEN BINDING
Antibody Specificity: The Key to an Antibody’s Usefulness Across Techniques
Too often, it is presumed that antibodies recognize the target antigen to which they were raised in any application and bind only that unique antigen across various species and experimental situations. Although these characteristics might be true for a given antibody, they are far from safe assumptions. The usefulness of an antibody is strictly linked to the availability of the epitope to which it binds in the technique being used, the presence of that particular binding determinant in other proteins within the sample (e.g., family members), and the conservation of the epitope across species. Relative to polyclonal antibodies (pAbs) recognizing multiple epitopes on the same target, monoclonal antibodies (mAbs) are less likely to be cross-reactive because they recognize one epitope, a single three-dimensional (3D) structure that provides a significant level of specificity. The advantage of specificity comes at the potential cost of mAbs not working across applications because that single binding epitope is not always available. pAbs raised against proteins or milieus that are more complex have the strength of recognizing several epitopes, which increases the chances of working in multiple techniques but also raises the likelihood of cross-reactivity with undesired targets. Thus, experimental controls are key to ensuring that the antibody only recognizes the target being studied. Controls for specificity include any sample that does not contain the antigen of interest (e.g., knockout and knockdown animals and cells), and natural sources in which the antigen is proven (e.g., by RT-PCR) not to be expressed.
Predicting in which assay an antibody will work is relatively difficult and is best addressed experimentally. Educated guesses can be made because epitope availability tends to correlate with the anticipated structure of a protein in the assay. When an antibody binds a relatively native protein conformation in one assay, it is likely to recognize it in another technique where the antigen is also native. For example, if an antibody works in a sandwich enzyme-linked immunosorbent assay (ELISA) with a relatively native form of the protein, it is also likely to bind the target in cell surface flow cytometry, immunoprecipitation, affinity purification, X-ray crystallography, and possibly even in vivo, assuming that there are no protein–protein interactions or target protein modifications that block or alter the antibody-binding site(s), respectively. Antibodies against extreme amino- or carboxy-terminal regions often work across many platforms irrespective of antigen conformation because they are usually accessible on the surface of the protein and usually lack significant higher-order structure. Antibodies that work in Western blots, where the protein has been denatured, are in many cases not the best candidates for applications in which the protein should be properly folded. Seeking antibodies that work well in techniques such as immunochemistry, immunofluorescence, and intracellular flow cytometry, where epitopes can be masked by protein–protein interactions or altered by the sample preparation process, is more problematic. Antigen retrieval techniques have the potential to reveal these otherwise concealed epitopes. Although pAbs tend to be the best first approach in these applications because they often bind multiple epitopes in the protein and thus have more chances to bind the target, many mAbs do work well and should not be discounted, especially because they can be much more specific. Ultimately, empirical evidence is the only definitive way to know if an antibody will bind the target in a particular application.
Antigenic Epitopes: The Antibody Docking Site
Usually, little is known about the antigenic determinants—the epitopes—that define the specificity of an antibody, unless the immunogen is a peptide. This is because the time and cost associated with epitope mapping often outweigh the benefits. Nevertheless, well-characterized epitopes can provide insights into the target antigen, particularly with respect to how it functions (e.g., recognizing an activated state of a protein) (Qi et al. 2012). Knowing where antibodies bind also allows one to evaluate cross-reactivity computationally. Even if it is not critical to establish the epitope for a specific line of experiments, understanding how epitopes are defined and identified drives a more informed use of antibodies. Excellent conceptual reviews (Morris 2007, 2008) and detailed protocols (Heckels and Christodoulides 2001; Pinilla et al. 2001; Rodda 2001; Freund et al. 2009; Reineke and Schutkowski 2009; Schaefer et al. 2010) are available for a more in-depth understanding of this topic, step-by-step procedures, and relevant troubleshooting considerations.
Different paratopes—the antigen-binding surfaces of antibodies—have the capacity to bind a broad range of structures. These range from small molecules, referred to as haptens (e.g., phosphocholine and vitamin K1), to more complex structures within polysaccharides and proteins (see Chapter 4 in Murphy 2012). Regardless of the complexity of an epitope, its structure is complementary in nature to that of the corresponding paratope. This discussion focuses on proteinaceous epitopes, which are traditionally characterized as either conformational (discontinuous and assembled) or linear (continuous and sequential). Conformationally dependent epitopes consist of specific amino acids from various, and often distant, positions along the primary protein sequence that are brought into close proximity and specific orientation in the tertiary protein structure. In contrast, linear epitopes are composed of residues that are contiguous in the primary protein sequence and are usually, but not exclusively, accessible in techniques in which the protein has been denatured. Thus, immunoblots are often used to determine if epitopes are more likely to be conformational or linear because the native 3D conformation is lost when the protein is denatured.
Epitopes have also been classified based on the complementary technical methodologies used to define them: “structural” and “functional.” The former relies on traditional structural biology techniques, whereas the latter delineates key residues based on experiments that measure the binding characteristics of the antibody. Structural and functional methods can identify the exact amino acids involved in antibody–antigen binding, narrow down the region in which the antibody binds the target, or define the epitope relative to some other portion of the protein (e.g., that of another antibody, a ligand, or a DNA-binding site). High-resolution structural studies suggest that, on average, 15–20 amino acids of the antigen are present at the paratope–epitope interface; functional techniques can narrow this down to four to eight residues that play a significant role in establishing a measurable antibody–antigen complex.
X-ray crystallography provides the highest degree of structural detail: It not only visualizes the amino acids that create the surface of the epitope that lie within the antibody’s antigen-binding pocket, it also implicates key residues necessary for binding. However, this method is seldom used because of the time, cost, and expertise needed to generate high-quality crystals and analyze the data. Nuclear magnetic resonance circumvents the need to generate crystals, but provides less resolution of the epitope and is limited to studying antibodies bound to small antigens, antigen fragments, and peptides.
Functional epitope mapping methodologies can be grouped into several categories: those that involve competition for antibody binding, approaches that involve antigen fragmentation or modification, and the use of peptides to define linear epitopes. Competition studies are relatively straightforward and define epitopes in relation to another binding site by asking if an antibody can bind after something else has bound the target (Fig. 1). This approach is extremely useful when looking for antibodies that simultaneously bind the same protein or protein complex, an essential characteristic of the antibodies used in sandwich ELISAs and protein arrays. It is also useful for finding antibodies that block ligand binding or prevent other complex formations with the antigen. When the antibody being studied and the other molecule do not simultaneously bind the target, one can infer that their respective binding sites are minimally close enough to interfere sterically with the binding of each other; or maximally that they are overlapping and possibility even identical. It is important to note that this assumes that binding of the first binder to the target protein does not alter the conformation of that protein significantly enough to eliminate the binding site of the second binder. This strategy is applicable to both conformational and linear epitopes but is limited to monovalent targets (i.e., one binding epitope per target molecule). Competition can be assessed by any technique in which antibody binding can be measured (e.g., ELISA, flow cytometry, immunoblots, and surface plasmon resonance). The main requirement is that one of the binders (e.g., antibody) be allowed to bind the antigen under saturating conditions, ensuring that all epitopes are occupied, before addition of the competing binder (e.g., a second antibody). To address the concern of a higher-affinity antibody displacing a lower-affinity one on some portion of the targets, the assay should be performed in both directions: In Sample A, Antibody A binds first; in Sample B, Antibody B binds first. If the results of these competition experiments do not agree, then displacement needs to be considered.
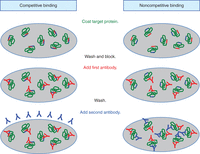
Binding competition assays. Binding competition assays can be used to assess if the epitopes of two different antibodies are in close proximity to each other, or possibly identical. First, one antibody is allowed to bind under saturating conditions. Second, the other antibody is added to see if it can bind as well. The illustration here depicts a standard ELISA, but this can also be executed with a sandwich ELISA format. When antibodies compete for the same region of the antigen, the presence of the first antibody precludes binding of the second. In contrast, noncompetitive antibodies can both bind because the epitopes are far enough apart.
Determination of an antibody’s ability to bind discrete antigen fragments can narrow down the region in which the epitope lies. By default, if an antibody binds to a particular fragment and not others, then the epitope must reside within the stretch of amino acids that makes up that portion of the target. This is useful for mapping epitopes that are not dependent on proper protein folding or when protein domains can be separated in a manner that preserves their overall tertiary structure. Antigen fragments can be obtained by recombinant expression or created via proteolytic digestion (Mazzoni et al. 2009).
Another general approach to obtain epitope information is to measure the ability of an antibody to bind a modified antigen (e.g., chemical modification, proteolysis, natural or recombinant mutants). A very detailed epitope map can be obtained using random mutagenesis (e.g., error-prone PCR, and mutator Escherichia coli strains) to generate small mutant libraries of the original antigen. Screening clones using ELISA, colony blotting, phage display, or another method can identify nonbinders, which can then be sequenced to determine the altered amino acid (Chao et al. 2004). Instead of random alterations, alanine-scanning libraries can assess the importance of individual amino acids within the primary sequence by substituting them with a simple alanine residue (Schrohl et al. 2011). Once regions containing disruptive changes point to the putative epitope, more targeted mutagenesis analysis can be undertaken to evaluate the involvement of specific residues in antibody binding. The strength of a mutagenesis approach is that the epitope can be mapped to specific amino acids, providing very high resolution. Although advances in recombinant DNA technologies continue to improve the utility of these approaches, creating and expressing the mutant libraries are often quite laborious and resource-intensive.
A significant caveat of all these techniques is that any lack of antibody binding might not be caused by changes in the epitope itself, but rather by changes in a region outside the antibody-binding site that induce a conformational change in the epitope and prevent antibody binding. In other words, the inability of an antibody to bind might be a secondary effect. Antibody “footprinting” (i.e., mapping by proteolysis or acetylation protection) does not suffer from this concern because, rather than modifying the antigen and determining if the antibody can bind the altered form, the antibody is bound to the antigen before modification to see which part of the antigen is protected by the antibody from modification (Jemmerson 1996).
Another strategy to narrow down the possible location of linear epitopes within a target is with overlapping-peptide libraries. Ideally, the overlapping peptides will be 15- to 20-mers that are offset by one residue, although the offset can be greater. Creating peptides such that each residue of the antigen is the starting point for a new peptide maximizes the chance of finding one or more peptides that bind the antibody. Comparing antibody binding to several truncated versions of the peptide(s) to which the antibody binds can delineate the minimum requirements of the epitope. Peptide libraries generated to measure how much a particular amino acid contributes to antibody binding are referred to as “scanning” libraries (Reineke 2009). As with the recombinant protein option noted above, alanine scan libraries systematically replace each residue in the peptide that the antibody binds with an alanine, whereas positional scan libraries change the residue at a set position to all possible amino acids. The latter is often used to see how a higher-affinity interface can be engineered. Including mutants containing two or three simultaneous changes can increase the complexity of the latter approach and ideally maximize the chances of finding a better epitope. Completely random peptide libraries are very useful in identifying linear epitopes when the antigen is unknown. Knowing what the natural epitope could contain can help researchers identify the antigen by searching protein sequence databases for that sequence.
Peptides can be synthesized or recombinantly displayed at the end of a protein expressed on phage, bacteria, or yeast. Traditional methods of synthesizing peptides require expensive instruments and can be unnecessarily costly when making peptide libraries. Fack et al. (1997) compares mapping via phage display and peptide libraries head to head. Thankfully, antigen-binding studies do not require milligram quantities, and a more economical option is available. Microgram quantities of many peptides can be generated on polystyrene “pins” in a 96-well-plate format. Antibody binding can be accessed directly on the “pins,” or the peptides can be released into solution (Rodda 2001) for studies evaluating their ability to block antibody binding.
Many functional epitope-mapping techniques require the antigen to be labeled or bound to a solid phase (e.g., the surface of a well in an ELISA plate) to detect binding. This runs the risk of changing the conformation of the antigen and, in turn, the availability of the epitope. It is also true for the antibody but is less of a concern given their general flexibility and resilience. This can be addressed by the increasing availability of highly quantitative, label-free methods, specifically surface plasmon resonance (SPR), biolayer interferometry (BLI), microcalorimetry, and resonant acoustic profiling. In most cases, analogous label-free versions of the assays described above can be constructed on these systems. Some of these approaches are discussed in more detail in the following section addressing affinity determination.
Complementarity Determining Regions: The Unique Characteristic of Each Antibody
The heavy- and light-chain variable domains of an antibody contain complementarity determining regions (CDRs) that endow antigen-binding specificity to the antibody. Because the amino acid sequence of each CDR is unique, it can be considered the antibody’s “fingerprint.” There are several benefits to determining this distinctive characteristic. By understanding the nature and diversity of paratopes specific for disease-associated antigens, rationally engineered therapeutic antibodies can be developed. Humanized antibodies and their recombinant derivatives, such as scFvs and bispecific antibodies, can be constructed (Yu et al. 2008; Demeule et al. 2009) based on rodent mAbs proven to have a desired in vivo activity in preclinical studies. In the former, CDRs of nonhuman origin are engineered into the context of the variable region of a human immunoglobulin backbone to minimize antigenicity to the foreign protein sequence (Almagro and Fransson 2008). Moreover, determining the CDR composition of antibodies produced by patients with autoimmune disorders when the antigens are not known has the potential to facilitate a more detailed diagnosis and opens the door to personalized medical treatment (Somers et al. 2009). Finally, the precise CDR sequence of an antibody is required for legal filings related to intellectual property protection.
Although many companies offer CDR sequencing, this can be performed easily in laboratories with basic molecular biology capabilities because it uses the same set of techniques used to sequence any protein expressed by a cell. Basically, mRNA isolated from the hybridoma is reverse-transcribed into a cDNA library. The specific variable domain cDNAs are amplified, cloned, and sequenced. Degenerative primers that enable heavy- and light-chain variable domain amplification from the species of origin are the key reagents in this common process. Such primers, as well as detailed protocols, are described in the literature for mouse and rat (Breitling and Dübel 1998; Toleikis et al. 2004; Siegel 2009; Strebe et al. 2010), rabbit (Rader 2009), llama (Harmsen et al. 2000), and human (Marks and Bradbury 2004; Ozawa et al. 2006; Tiller et al. 2008; Andris-Widhopf et al. 2011) variable regions. Online tools (http://www.abysis.org/; http://www.imgt.org; http://www.vbase2.org) exist to facilitate alignment with existing immunoglobulin databases and to delineate the CDRs within the variable regions. More extensive discussion of the alignment and analysis process and relative merits of various approaches can be found in Martin (2010).
Affinity: The Strength of an Antibody–Antigen Complex
The usefulness of an antibody is greatly influenced by the strength of its noncovalent bond to its antigen. High-affinity antibodies are key for detecting low-abundance targets and in assays in which the antibody–antigen complex must endure extensive washing. Comparatively lower-affinity antibodies can be better options for affinity-purification techniques because they allow elution of the target under milder conditions. Antibody affinity constants span at least seven orders of magnitude, stretching from submicromolar to beyond picomolar affinities (Harlow and Lane 1999). Techniques measuring the equilibrium between the antibody–antigen bound complex and the free molecules can be as basic as equilibrium dialysis and ELISAs or as sophisticated as SPR, BLI, microcalorimetry, and resonant acoustic profiling. This section provides a brief overview of the first four.
Affinity constants for antibodies whose targets are small can be obtained using equilibrium dialysis (Kariv et al. 2001; Murphy 2012), which leverages the size difference between the partners and their differing abilities to cross a dialysis membrane. A standard experiment involves separating equal volumes of buffer with dialysis membrane through which antibody cannot cross but the antigen can (e.g., 10 kDa cutoff). The overall process is as follows: Side A starts with a fixed amount of antibody and Side B with a known concentration of the respective antigen. As free antigen diffuses across the membrane, a portion of molecules is bound by the antibody and consequently sequestered on Side A. Two equilibrium states are established over time: (1) bound antigen versus free antigen and (2) free antigen on Side A versus Side B, which becomes equal because it can freely cross the membrane. In samples in which a control antibody is used, the free antigen concentration on both sides of the membrane becomes half that of the starting concentration because the antibody does not deplete the system of free antigen. In contrast, the antibody being tested reduces the concentration of free antigen by binding it and is dependent on the affinity of the antibody for its target as well as the molar amounts of antibody and antigen used. This simple design is repeated over multiple dilutions of antigen while keeping the antibody concentration constant. The best range of dilutions needs to be determined empirically. The amount of antigen bound to the antibody is equal to the total amount of antigen added to the system less that amount of antigen remaining free once equilibrium has been established. Affinity constants are determined by plotting bound versus free antigen across different antigen concentrations and analyzing these data points using nonlinear regression (Leatherbarrow 1990). The work involved in setting up many dialysis conditions is less onerous using commercially available systems (e.g., 96-Well Equilibrium DIALYZER and Multi-Equilibrium DIALYZER from Harvard Apparatus).
ELISA is another sensitive and inexpensive method to measure antibody affinity. Relative affinities—basically, how much better does Antibody A bind Antigen X relative to Antibody B—can be determined using ELISA by measuring how much each antibody can be diluted before significant signal is lost. When each antibody starts at the same concentration, the one that can be diluted the most has the higher affinity. The assumption is that each antibody binds only one epitope per target. Because the amount of antigen coating the bottom of the well is unknown and, in turn, the amount of antibody bound to the antigen is unable to be determined, exact affinity constants are unattainable with this approach. An additional drawback of this method is that the 3D structure of the antigen can change on binding to the plastic. To obtain an affinity constant of a completely unmodified or altered antibody–antigen complex, the ELISA can be designed to measure the amount of free/unbound antibody left over after binding to the antigen in solution (Heinrich et al. 2010; Martineau 2010). This approach requires preincubating the antibody with the antigen until equilibrium is reached, then transferring the antibody/antigen mixture to an antigen-coated ELISA plate to determine the amount of free/unbound antibody:
-
1. Coat the ELISA plate in which free antibody is to be measured with antigen.
-
2. Block the plate.
-
3. Add the equilibrated antibody–antigen mixtures to the coated wells and incubate.
-
4. Wash away antibody–antigen complexes as well as free antibody that has not bound to the well.
-
5. Incubate with secondary antibody labeled with horseradish peroxidase to detect the previously free antibody that is now bound in the well.
-
6. Wash away unbound secondary antibody.
-
7. Develop and read on an ELISA plate reader.
-
8. Extrapolate free antibody concentrations from a standard antibody curve run in parallel on the same ELISA plate.
It is critical to have just enough antigen coated on the ELISA plate used to measure the amount of free antibody in the antibody–antigen mixture, such that the equilibrium between free and bound antibody is not altered during the incubation of the mixture in the antigen-coated ELISA plate. When the free/unbound antibody in the antibody–antigen solution begins to bind the antigen coated in the well, the free antibody is depleted from the solution; this shifts the equilibrium between the free and bound antibody in solution, distorting the affinity measurements. The goal is to have enough antigen bound to the plate to capture as little free antibody as possible while still being able to generate a readable signal, but not so much as to significantly deplete the free antibody from the antibody–antigen solution being tested and skew the affinity data. The process of establishing the best antigen coating concentration is explained in Chapter 41 of Antibody Engineering (Martineau 2010) and involves (1) incubating various dilutions of the antibody (without antigen) in wells of an ELISA plate coated with the antigen; (2) transferring the “depleted” antibody dilutions from the original plate to a second ELISA plate coated at the same time and under the same conditions as the original one; (3) incubating the “depleted” antibody dilutions in the second ELISA plate for the same amount of time as the original; and (4) processing both plates as you would for an ELISA (wash/incubate with secondary antibody/wash/develop). If the plots from the original (first) and second ELISAs are almost identical, then very little antibody was “depleted” during the original incubation. The goal is to have the slopes of the lines obtained by plotting absorbance versus antibody concentration in both plates be within 10% of each other. When the difference is >10%, this sequential ELISA needs to be repeated with plates coated with less antigen.
The other important variable that needs to be established during this optimization process is the antibody concentration. The concentration of antibody used in the final affinity determination assay needs to be twice that of the highest concentration that still produced readings within the linear range of the optimization assay (not saturated). Knowing the antigen coating concentration and the concentration of antibody that will be allowed to prebind with various amounts of antigen in solution, one can begin the ELISA experiment from which the affinity will be determined. A nonlinear regression analysis should be applied to the data, in which absorbance (a measure of how much free antibody existed after the antibody–antigen equilibrium was established) is plotted against the solution antigen concentration. This experimental approach can measure up to nanomolar affinities and be modified to obtain rates of binding by varying the time of incubation rather than the concentration of the antigen in solution (Hardy et al. 1997; Zhuang et al. 2001).
Many methods for interrogating biomolecular interactions depend on fluorescent, enzymatic, or radioactive labels. When attached to the primary interactors (e.g., the antibody being probed), these labels risk altering the interaction and impacting the measured values. Even when incorporated only as indirect labels (e.g., enzyme-labeled secondary antibodies that then bind to the antibody being evaluated), unaccountable variables can be introduced because detection is based on the behavior of the label, not the target molecule(s). Surface plasmon resonance and related techniques are “label-free” methods for macromolecular kinetic analysis and are now at a point at which assay development time is minimal and relatively little expertise is required to use many of these systems effectively. There continues to be a fairly rapid expansion in this area, so this discussion considers only a few of the most widely available approaches.
The first experimental paradigm involves using optical techniques to measure macromolecular binding events quantitatively. This category includes surface plasma resonance (e.g., GE Biacore and SensiQ Pioneer), biolayer interferometry (e.g., FortéBIO Octet), and resonant waveguide grating (RWG; e.g., Corning Epic and PerkinElmer EnSpire), all of which measure dynamic changes in optical refractive indices across time and under various binding and nonbinding conditions to calculate the affinity two molecules have for one another. Typically, a homogeneous pool of one of the potential interactants (e.g., antibody) is used to coat a biosensor surface; a variety of functionalized biosensor surfaces (Protein G, nickel–NTA, streptavidin, etc.) are available on which the unmodified first interactant can easily be immobilized. Under controlled conditions, the coated biosensor is then exposed to a second free unlabeled interactant (e.g., antigen). Binding of the partner molecules to each other leads to dynamic changes at the surface of the biosensor, which are then detected by the system. These changes are used to calculate the binding characteristics of the interactants directly. It is important to note that kinetic determinations are limited to molecules that fall within the appropriate size ranges of detection for the particular instrument (see Fang (2006) for a broader discussion of optical techniques).
SPR was developed in the early 1990s by the Swedish company Biacore AB (now part of GE Healthcare) and is the most well-established technology in this area owing to its long-standing availability. This technology has been incorporated into many other systems (e.g., Bio-Rad ProteOn, SensiQ Pioneer, and Reichert SPR System). Consequently, it has become the de facto benchmark for the field, and more extensive comparisons have been performed with orthogonal technologies. Thus, a major advantage of SPR use is the wealth of studies available that examined antibody–antigen kinetics with SPR-based instruments (e.g., Vancott 1999; Williams et al. 2007; Reddy et al. 2012).
Biolayer interferometry represents one of the most prevalent alternatives to SPR (Krause et al. 2011; Kim et al. 2012) and uses fiber-optic biosensor technology. Advantages of BLI include shorter experimental run times, reduced sensitivity to refractive index fluctuations of the sample, and the lack of dependence on microfluidics.
Resonant wave grating (Epic from Corning) uses patterned surfaces to reflect distinct wavelengths when illuminated by broadband light. These wavelengths vary in response to mass changes caused by molecules moving into and out of the measurement region. This information is then used to calculate binding strength. A significant advantage of the technology is the ability to measure interaction strength in both biochemical and cell-based assays. Unfortunately, it is only able to measure binding strength and cannot establish actual on- and off-rate kinetics, which the SPR and BLI systems can.
Although there are variations between optical biosensor methods with respect to format, use of microfluidics, and their precise detection modalities, they do share basic design features. As such, many of the same considerations apply to all these systems. First, low surface density of the molecule bound to the biosensor surface is essential to avoid so-called mass transfer limitations, which can lead to falsely low dissociation-rate-constant determinations. Basically, molecules on the surface need to be sufficiently spaced such that the free analyte is able to diffuse into the bulk solution before it is bound by another interactant on the surface (e.g., another antibody). Accurate off-rate calculations also require the molecules to bind and dissociate in a detectable fashion. If they do not, the instrument will read this as a higher-affinity interaction than it actually is. A similar effect can stem from a very fast binding interaction between the molecules that effectively outpaces the rate of analyte diffusion in from the bulk solution. Another standard recommendation is to set up the assay with the antibody coupled to the sensor and the antigen as the analyte in solution. There are a variety of options for immobilizing antibodies via the constant regions to the biosensor that do not interfere with antigen interaction. An additional advantage is that biosensor regeneration is relatively simple when the antibody is bound to the sensor because they are very resilient proteins, whereas regeneration conditions would need to be carefully determined for each new antigen-based biosensor. When designing assay conditions, it is also critical to include appropriate negative controls to account for nonspecific interaction effects. In general, the ideal control here is a biosensor coated with a protein known not to interact with the analyte being assayed. This allows one to make the best compensatory adjustment for background analyte binding. An alternative, or parallel, control is the inclusion of negative biosensors. This typically includes both a blank biosensor (no molecule attached) run against the standard analyte containing buffer and a loaded biosensor measured against analyte-free buffer. Inclusion of an appropriate blocker reagent to reduce nonspecific binding should also be determined. The type of blocker varies according to the chemistry of the biosensor. For example, biocytin is a good choice for blocking streptavidin-based biosensors after the interactant has been bound. Finally, each system has its own unique methodology for data quality assessment and extrapolation. Understanding and strictly following these guidelines is critical for proper interpretation of the relatively complex data sets generated.
The optically based methods above are hardly the only means of analyzing antibody/antigen interactions. Many other methods have also been used to analyze these and other molecular interactions such as isothermal calorimetry (ITC; Van Antwerp and Wittrup 1998), analytical ultracentrifugation (AUC; Demeule et al. 2009), and backscatter interferometry (Bornhop et al. 2007), to name a few. Although there is little published data systematically comparing the performances of these varied approaches (ELISA, optical methods, ITC, etc.), the few available studies suggest that the affinity constant estimations are similar across platforms (Myszka et al. 2003; Wartchow et al. 2011), whereas another study comparing ELISA with SPR concluded that they are not only consistent but also complementary (Heinrich et al. 2010). This latter study is interesting because the ELISA distinguished two distinct binding populations in solution (high- and low-affinity constants), but the affinity constants obtained via SPR were dependent on whether binding occurred in solution or at the surface of the biosensor: A higher-affinity constant was detected under solution binding conditions and a lower one when the antibody was bound to the surface or the Biacore chip.
THE ANTIBODY BACKBONE
Isotype: The Constant Regions
The majority of the heavy chain and approximately half of the light chain consist of constant domains, which define an antibody’s particular isotype. In the mouse, the heavy-chain isotypes are IgA, IgD, IgE, IgG, and IgM, whereas the light chains exist in two forms, kappa and lambda. IgGs are further subdivided into subclasses IgG1, IgG2a, IgG2b, and IgG3. Humans have slightly different isotype subclasses for IgG (IgG1, IgG2, IgG3, and IgG4) and have two IgA subclasses (IgA1 and IgA2).
Determining the isotype of a monoclonal antibody derived from mouse or rat is rather easy given the plethora of commercially available kits (BD Biosciences, Roche Applied Science, Sigma-Aldrich, Southern Biotech, Thermo Scientific Pierce). These kits typically use sandwiching of the sample antibody between two anti-isotype secondary antibodies. The formats vary in level of complexity, execution time, equipment needed, and cost per sample tested. The most common are isotype “strips” and ELISA-based kits. BD Biosciences also offers a FACS-based approach (BD Cytometric Bead Array, Mouse Immunoglobulin Isotyping Kit). Unfortunately, the reagents to determine isotypes from hamster and rabbit mAbs are not readily available. Spurious identification of more than one isotype complicates the interpretation of these assays, with the isotype strips being the most susceptible and the FACS approach being reported as the least likely to be plagued by this. When the test scores positive for more than one isotype, diluting the test material can often eliminate this mixed result. Protocol: Isotype Determination of Rodent-Derived Monoclonal Antibodies Using Sandwich ELISA (Weis-Garcia and Carnahan 2017) outlines the ELISA-based isotyping process.
Isotyping can also be performed using the assay in which the antibody is being used. Rather than capturing the unknown antibody with an antibody, the unknown can be captured using the target antigen in an ELISA, on cells for flow cytometry, on a membrane for immunoblotting, etc., and probed with a different isotype-specific antibody. This approach allows one to isotype an antigen-specific mAb even when other antibodies that do not bind the antigen are present. This can be very useful when screening for new mAbs after a fusion when the goal is to identify and preserve hybridomas secreting antigen-specific antibodies of various IgG isotypes. If the initial screening uses an anti-Ig secondary antibody to identify antigen-specific secretors quickly, the next level of screening can use anti-IgG1-, IgG2a-, IgG2b-, and IgG3-specific secondary antibodies to further identify the cultures worth preserving. If having a specific isotype is central to the intended application, screening for other isotypes is necessary when one isotype is dominant (as IgG1 often is).
Effector Domains: The “Other” Biological Activity
For decades, researchers have harnessed the ability of antibodies to identify and locate their respective antigens. This, however, is only half of what antibodies can do. The “other” biological activities reside opposite the antibody binding arms in the Fc domain, which is composed of heavy-chain constant regions after CH1. Collectively, Fc domains focus and stimulate the humoral arm of the adaptive immune system, mediate antibody transport through cells, and ensure healthy IgG levels in the blood. All of these functions are accomplished by binding different proteins, specifically Fc receptors and complement C1q. Here, we provide an overview of various biological activities elicited by different human isotype groups and the binding proteins essential for Fc-driven actions (Table 1). A deeper appreciation for this complex biology can be obtained by perusing Johansen et al. (2001), Kraft and Novak (2006), Woof and Kerr (2006), Nimmerjahn and Ravetch (2007), Roopenian and Akilesh (2007), Aschermann et al. (2010), Schroeder and Cavacini (2010), Chen and Cerutti (2011), Desjarlais and Lazar (2011), and Murphy (2012).
Human isotype classes and associated Fc-binding proteins
IgAs exists in two forms, monomeric and dimeric. The main protective role of this isotype is the neutralization of bacteria, viruses, and toxins. Although the Fc domain is not necessary for neutralization per se, it is essential in transporting dimeric IgA to locations where this functionality is needed but the antibody is not made. The mucosal surfaces of the intestinal and respiratory tracts are a prime target for the deleterious actions of pathogens and toxins, but they do not contain B cells to produce protective antibodies. When the Fc domain of the dimeric IgA, associated via the J (joining) chain, binds the polymeric immunoglobulin receptor (pIgR), it is transcytosed across the epithelium into the mucosal lining of these tracts. IgA is also present in glandular secretions such as breast milk, saliva, and tears, but transcytosis into these liquids is mediated by the neonatal Fc receptor (FcRn). IgA resides in the extracellular tissue spaces and in the blood, where it not only neutralizes invaders but also promotes their destruction by antibody-dependent cell-mediated phagocytosis (ADCP) through the IgA Fc receptor (FcαR, FcaR, and CD89).
IgD is the least well-understood isotype. It is found in mucosal secretions and at low levels in the blood. Binding of the constant IgD region to particular bacterial proteins activates B cells. To date, no IgD FcR or binding protein has been identified on effector cells. If IgDs function in a manner similar to the other isotypes, their Fc domains must bind the putative FcdR on the surface of basophils, because cross-linking IgD leads to release of immune-activating, proinflammatory, and antimicrobial factors from these effector cells.
IgGs are the most versatile isotype because the Fc domains of the various subclasses endow this class as a whole with the broadest spectrum of biological activity. They activate complement, modulate effector cell functions, and enable the antibody to undergo transcytosis. The FcRn is the binding protein responsible for transporting IgGs’ protective immunity across the placenta into the blood stream of the fetus in utero. Unlike the pIgR that essentially transports in one direction, the FcRn shuttles IgG back and forth across the epithelia that line the gut. Binding of the IgG Fc domain to FcRn also protects the antibody from degradation and excretion by recycling it back into the circulatory system. This function is key to maintaining healthy IgG levels in the circulatory system. IgGs also bind more traditional, effector cell–expressed FcRs: Fcγ-RI (Fcγ receptor I), FcγRII-A, FcγRII-B1, FcγRII-B2, and FcγRIII. Given the multiple IgG subclasses and several FcγR types that collectively activate and inhibit effector functions, IgGs orchestrate their arm of the humoral immune response quite elegantly. Antibody-dependent cellular cytotoxicity (ADCC) is mainly performed under the direction of IgGs. Antigen–IgG–FcγR complexes not only target small pathogens for destruction by effector cells via ADCP, but they also steer the release of lysosomal and secretory granule components to attack larger parasites such as worms that cannot be engulfed by phagocytes. Although much is known about how all four IgG isotypes and five FcγRs work together to tightly regulate these effector cell functions, complete appreciation for this finely tuned system is still being sorted out. IgGs also fix complement and initiate the classical cascade when they coat the surface of a pathogen expressing an antigen they recognize (see Chapter 2 in Murphy 2012). It is important to note that unlike the other antibody isotypes, the glycosylation state of IgGs significantly impacts how well it mediates effector function, presumably because of its role in antibody–FcγR binding; when the only glycosylation site on the IgG heavy chain is mutated, the “sugarless” protein shows minimal to no affinity for the FcγR. These in vitro studies suggest an in vivo role for IgG glycosylation, which is supported by the fact that the glycosylation site—an asparagine near the hinge regions of the Fc domain—is evolutionarily conserved. Furthermore, effective ADCC and complement-dependent cytotoxicity (CDC) require properly glycosylated IgG. The dependence of IgGs on this posttranslational modification is likely because the sugar moiety is sequestered between the Fc arms of each heavy chain in a hydrophobic pocket, which is not true of the other antibodies (Lux and Nimmerjahn 2011).
IgEs mainly function to initiate the inflammatory response, and they have a very high affinity (0.1 nm) for the FcεR1 receptor even in the absence of antigen. Engagement of multivalent antigens with the prebound IgE results in mast cell degranulation, releasing granule contents such as histamine, serine esterases, proteases, cytokines, chemokines, and lipid mediators into the local extracellular space.
IgMs are mainly found in the circulatory system, but a portion is relocated into the lumen of the gut via the pIgR, the same receptor that transports dimeric IgA across epithelium into mucosal surfaces. Because these are the only two isotypes transcytosed by pIgR and associated with the J chain, it is not surprising that the J chain is essential for their pIgR-mediated epithelial transport. The key role of IgM is to initiate the classical complement cascade by binding complement component C1q to its Fc domain only after it has opsonized the target by forming immune complexes with the respective antigen covering the pathogen’s surface. IgM activates complement more efficiently than IgG because it only takes one IgM antibody to activate one molecule of C1q, whereas it takes at least two IgGs that are 30–40 nm apart on the outside of the pathogen to bind a C1q and trigger the cascade.
Footnotes
-
From the Antibodies collection, edited by Edward A. Greenfield.








