
Analysis of DNA Methylation in Mammalian Cells
Abstract
Methylation of DNA, the most experimentally accessible epigenetic alteration of eukaryotic cells, has generated an extensive literature and an abundance of analytical tools. The term “methylome” (referring to the complete set of cytosine modifications in a genome) is appearing with greater frequency in the literature, reflecting the growing number of researchers in the field. Here we introduce a set of robust protocols for methods that can be performed routinely for the elucidation of DNA chemical modifications involving methylation of cytosine. The strengths and limitations of each approach are also discussed.
DNA METHYLATION AFFECTS AND REVEALS BIOLOGICAL PHENOMENA
In adult mammals, ~40% of CpG dinucleotides in the genome contain methylation marks at the 5′ position of cytosine. A large proportion of methylation marks are erased during germ cell development, and an additional wave of demethylation of the incoming paternal genome occurs in zygotes. The reestablishment of DNA methylation marks begins early in embryonic development and is largely complete at the time of birth. During differentiation, distinct subsets of methylation marks are established and become heritable in different somatic cell lineages, constituting a stable memory of developmental commitment during cell division. Among the most dramatic manifestations of this memory are X-chromosome inactivation and genomic imprinting, both of which are associated with stable, chromatid-specific silencing of gene loci. Somatically heritable DNA-methylation-mediated silencing also occurs in vast regions of the genome containing interspersed repetitive elements. In promoter-associated clusters of methylated cytosines, known as “CpG islands” (see Box 1), DNA methylation stabilizes condensed chromatin states initiated by binding of Polycomb group protein complexes. Histone modifications occurring at these sites recruit DNA methyltransferases, and, acting together, these interacting proteins can propagate multiple methylcytosine marks over an entire CpG island. These chemical changes affecting the DNA sequences near each promoter represent a layer of epigenetic control of a specific chromatin state that is somatically heritable, but nonetheless remains susceptible to somatic reprogramming. Loss of subsets of specific methylation marks occurs during normal development, as well as during inflammatory or pathological processes. DNA methylation can be modulated in the absence of cell division. Notable examples are a region in the promoter–enhancer of the interleukin-2 gene, which is demethylated in T lymphocytes following activation (Bruniquel and Schwartz 2003; Murayama et al. 2006), as well as demethylation and transcriptional activation of the neuronal plasticity gene reelin during contextual fear conditioning in the hippocampus (Miller and Sweatt 2007). The latter example emphasizes the fact that DNA methylation marks are dynamic and highly responsive to environmental factors.
PUBLIC DOMAIN SOFTWARE FOR IDENTIFYING CpG ISLANDS IN PROMOTER AND CODING REGIONS OF MAMMALIAN GENES
Several programs are freely available over the Internet that simplify the task of identifying CpG islands. Some of these programs are listed in Table 1. Here we describe briefly the two computational methods that are used most frequently [at the time of this original writing] for the prediction of CpG islands (CGIs).
CpGcluster CpG Island Prediction
CpGcluster (https://github.com/bioinfoUGR/cpgcluster) is a fast and computationally efficient algorithm that uses only integer arithmetic to predict statistically significant clusters of CpG dinucleotides. All predicted CGIs start and end with a CpG dinucleotide, which should be appropriate for a genomic feature whose functionality is based precisely on CpG dinucleotides. The only search parameter required in CpGcluster is the distance between two consecutive CpGs. None of the statistical properties of CpG islands (e.g., G+C content, CpG fraction, or length threshold) that are often required in other programs are needed as search parameters, which may lead to the high specificity and low overlap with spurious Alu elements observed for CpGcluster predictions (Hackenberg et al. 2006, 2010).
HMM Model-Based CpG Island Prediction
The Irizarry laboratory developed software (makeCGI) that uses a hidden Markov model (HMM)-based approach to CpG island prediction. They have fit the HMM model to genomes from 30 species, and the results are available at the University of California Santa Cruz genome browser (http://genome.ucsc.edu/goldenPath/customTracks/custTracks.html#Multi) (Irizarry et al. 2009; Wu et al. 2010). Their results support a new view toward the development of DNA methylation in species diversity and evolution. The observed-to-expected ratio (O/E) of CpG residues in islands and nonislands segregated closely along phylogenetic lines and shows substantial loss in both groups in animals of greater complexity, while maintaining a nearly constant difference in CpG O/E ratio between islands and nonisland compartments.
Programs for the identification of CpG islands
In summary, DNA methylation can be regarded as a metastable digital record of interactions between the genome and its environment that is stored in the form of a binary string of “zeros” (unmethylated bases) and “ones” (methylated bases). In the context of a rich diversity of responses to biological phenomena, the analysis of DNA methylation in mammalian cells can reveal aspects of developmental lineage history overlaid with changes induced by environmental influences as a cell- and tissue-specific record of an organism’s life history. Thus, measurement of methylation marks at the single base level can reveal developmental events, trace normal or abnormal cell lineages, document accidental environmental insults, and generate metrics of cellular aging (Shibata and Tavare 2006). However, genomic methylation is neither perfect nor static. Superimposed on acquired patterns of DNA methylation is noise generated as a result of the imperfect propagation of DNA methylation marks during semiconservative DNA replication. Occasional failures in the replication of the marks occur in dividing cells, and these stochastic variations constitute a biological clock recorded within cell lineages as binary strings in each cell’s DNA.
The Chemistry of DNA Methylation and the Bisulfite Reaction
The most common DNA methylation marks involve the addition of a 5′-methyl group to a cytosine that is part of a CpG dinucleotide, denoted as mCG. Less common, but widespread in stem cell progenitors, is 5′-methylation of cytosines in other contexts, denoted as mCHG and mCHH (where H is A, C, or T) (Lister et al. 2009). In addition, 5-hydroxymethylation of cytosine (hmCG) has been reported to occur frequently in neurons (Kriaucionis and Heintz 2009).
The “gold standard” for analysis of cytosine methylation at the sequence level is based on the modification of DNA with sodium bisulfite. This reaction exploits differences in the kinetics of deamination of cytosine and methylcytosine: Under the conditions used, cytosine is deaminated and converted to uracil more rapidly than is methylcytosine. When the deaminated DNA is amplified using the polymerase chain reaction (PCR), the newly created uracil residues in the template DNA then direct incorporation of adenosine in the synthesized strand. At the end of the PCR, the amplified DNA will contain a thymidine residue wherever unmethylated cytosine had been present, whereas cytosine will remain unchanged at those positions where the base was methylated in the original DNA sample.
EXPERIMENTAL APPROACHES FOR ANALYSIS OF DNA METHYLATION
In general, analysis based on DNA sequencing will provide superior resolution of modified bases; but in some instances, nonsequencing-based methods may be more cost effective. A general overview of available experimental approaches for analysis of DNA methylation is presented in Table 2 and Figure 1.
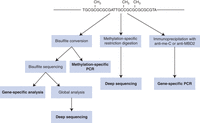
Methods for analyzing DNA methylation profiles in genomes. Step-by-step protocols are available for procedures in boldface (for gene-specific analysis, see Protocol: DNA Bisulfite Sequencing for Single-Nucleotide-Resolution DNA Methylation Detection [Lizardi et al. 2017a]; for methylation-specific PCR, see Protocol: Methylation-Specific Polymerase Chain Reaction (PCR) for Gene-Specific DNA Methylation Detection [Lizardi et al. 2017c]; for gene-specific PCR, see Protocol: Methyl-Cytosine-Based Immunoprecipitation for DNA Methylation Analysis [Lizardi et al. 2017d]; for deep sequencing after methylation-specific restriction digestion, see Protocol: High-Throughput Deep Sequencing for Mapping Mammalian DNA Methylation [Lizardi et al. 2017e]; and for deep sequencing after bisulfite conversion, see Protocol: Illumina Sequencing of Bisulfite-Converted DNA Libraries [Lizardi et al. 2017b]).
Overview of methods for DNA methylation analysis (at the time of this original writing)
Bisulfite Sequencing for Single-Base Resolution of DNA Methylation
The method most frequently used (at the time of this original writing) for analysis of cytosine methylation at the sequence level is based on modification of DNA with sodium bisulfite (Protocol: DNA Bisulfite Sequencing for Single-Nucleotide-Resolution DNA Methylation Detection [Lizardi et al. 2017a]). This approach has the advantage that every candidate cytosine can, in principle, be interrogated for its methylation status. A disadvantage is that bisulfite treatment always results in fragmentation of the DNA sample, limiting the analysis of sequence reads in a continuous strand of DNA to 800 bases, at best.
Targeted bisulfite sequencing typically involves a limited number of loci of interest, which are amplified using PCR. Because these PCR amplicons are usually analyzed by standard DNA sequencing, an interesting and lower-cost alternative is the EpiTYPER platform. EpiTYPER transcribes bisulfite-converted DNA in vitro into RNA molecules, which are subjected to base-specific cleavage followed by analysis of the fragments using mass spectrometry. EpiTYPER is partially automated, and thus this platform is well suited for analysis of medium-sized sets of CpG islands of interest (48–96) in studies involving large numbers of samples. For experimental questions that require whole-genome analysis of DNA methylation, DNA treated with bisulfite can be used to generate libraries suitable for sequencing in any of the major second-generation platforms (Roche 454, Illumina, or ABI SOLiD). Any of these sequencing platforms can generate genome-wide coverage, but the high cost of each global analysis experiment can be a limiting factor in deploying the technology. Protocol: Illumina Sequencing of Bisulfite-Converted DNA Libraries (Lizardi et al. 2017b) has been used by academic laboratories to analyze DNA methylation at the whole-genome level using the Illumina Genome Analyzer.
Methylation-Specific PCR for Gene-Specific DNA Methylation Detection
Methylation-specific polymerase chain reaction (MS-PCR) uses bisulfite-converted DNA as the starting material and conditionally generates DNA amplicons based on the use of two different sets of primers, one for methylated DNA and another for unmethylated DNA (Protocol: Methylation-Specific Polymerase Chain Reaction (PCR) for Gene-Specific DNA Methylation Detection [Lizardi et al. 2017c]). While not providing single-base resolution for the entire DNA amplicon, the method has the advantages of simplicity, flexibility, and low cost.
Immunoprecipitation of Methylated DNA Using Antibodies or Methyl-Binding Protein 2
Proteins or antibodies capable of specifically binding to DNA that contains 5mC are in principle capable of generating DNA fractions enriched in or depleted of methylated cytosines (Protocol: Methyl-Cytosine-Based Immunoprecipitation for DNA Methylation Analysis [Lizardi et al. 2017d]). These methods work best with single-stranded DNA and typically require relatively large DNA inputs. As pointed out (Laird 2010), the ratio of input DNA to affinity reagent can affect the enrichment efficiencies of genomic regions with varying 5mC density. Because the results obtained from immunoprecipitation methods are affected by the stoichiometry of the reagents, it is important to perform preliminary experiments to optimize and standardize the design of the capture experiments. After immunoprecipitation, a variety of analytical methods are available for generating DNA methylation data. [At the time of this original writing] immunoprecipitation followed by DNA microarray analysis (MeDIP) has been used in a large number of studies, and we expect that the use of immunoprecipitation in combination with high-throughput deep sequencing will become widely used in the future.
Global Analysis of DNA Methylation Using Restriction Endonucleases
Restriction endonucleases are powerful tools for assessing the methylation status of DNA. They have been used as the basis for locus-specific and genome-wide analysis of DNA methylation. Using a single endonuclease limits sequence sampling, but combinations of restriction endonucleases enable genomes to be fractionated into predominantly methylated or unmethylated compartments. An evaluation (Irizarry et al. 2008) of alternative microarray-based methods of DNA methylation analysis reported that approaches based on the use of the methylation-dependent endonuclease McrBC and an optimized bioinformatics approach for data analysis (CHARM) generated results with reasonably high correlation coefficients (0.76) when compared with data generated using the same samples in the Illumina Infinium HumanMethylation 27 BeadChip. Other methods evaluated in this study included MeDIP, HELP (Oda and Greally 2009), and McrBC (without CHARM data analysis); the correlation coefficients relative to the Infinium reference data set were 0.38, 0.48, and 0.63, respectively. No microarray-based methods are included here, because [at the time of this original writing] researchers increasingly feel that sequencing-based analysis is the preferred readout for analysis of genomic compartments generated by methylation-sensitive or methylation-dependent restriction endonucleases.
Protocol: High-Throughput Deep Sequencing for Mapping Mammalian DNA Methylation (Lizardi et al. 2017e) provides a method for genome-wide DNA methylation analysis (Edwards et al. 2010) that is based on enzymatic fractionation of the genome into methylated and unmethylated compartments. Because the method avoids the use of bisulfite modification, DNA fragments of relatively large size are preserved, which permits the generation of paired-end libraries with DNA inserts of known size in the ranges of 0.8–1.5, 1.5–3, and 3–6 kb. In most instances, the paired-end configurations can be uniquely mapped to the genome using software available from Applied Biosystems. This methodological approach delivers a reasonable balance between genome coverage and cost and is uniquely able to analyze the methylation status of repetitive elements in the human genome. Because it samples relatively long sequence domains, this method is also capable, in many instances, of generating valuable strand-specific information from imprinted regions in the mammalian genome.
High-Throughput Deep-Sequencing Technologies to Analyze Genome Partitions or Entire Genomes
Second- and third-generation high-throughput sequencing platforms are especially powerful when used for the analysis of bisulfite-converted DNA (see Box 2), because the availability of multiple reads for each locus in the genome reveals the fine structure of DNA methylation marks within cell populations. The richness of the information provided by deep sequencing is most dramatically apparent when sequencing reads are longer than 150 bases. Zeschnigk et al. (2009) have reported the DNA methylation status of more than 6000 CpG islands in human blood and human sperm samples. The methylation profiles of CpG islands revealed by this type of analysis often display the different methylation patterns in different reads of the same locus, indicative of imprinting-specific methylation or allele-specific regulatory patterns that remain to be elucidated.
POSTSEQUENCE PROCESSING OF HIGH-THROUGHPUT BISULFITE DEEP-SEQUENCING DATA
High-throughput deep sequencing generates terabytes of sequence data. These deep-sequencing technologies are fast, reliable, and relatively affordable platforms for obtaining genome-wide information. If the resolution is high enough, they can also provide information at single-nucleotide resolution. The technology presents several challenges to obtaining highly reproducible data, and, furthermore, the analyses are expensive to perform. Thus, high-throughput deep sequencing is not the method to use if a project requires only small-scale, low-resolution sequence information. In addition, owing to the lack of universal software for efficient and appropriate data management, the downstream processing and analysis of terabytes of data generated by each run become extremely difficult. Table 3 describes some of the commonly used software programs used in the analysis, alignment, assembly, and visualization of deep-sequencing data.
At the time of this original writing, there is no standard method for analyzing genome-scale high-throughput data obtained from deep-sequencing methodologies. The National Center for Biotechnology Information (NCBI) created a portal, called the Sequence Read Archive (SRA), which has been designed to meet the challenges presented by high-throughput deep-sequencing technologies. SRA provides a central repository for short read sequencing data and provides links to other resources referring to or using these data. It also allows retrieval based on ancillary information and sequence comparison. Finally, it establishes the basis for user-interactive submission and retrieval.
Summary of data and statistical analysis methods used by different genome-wide high-throughput deep-sequencing DNA methylation studies
Another advantage of high-throughput deep-sequencing approaches is their greater power for discovery of cytosine modifications that occur outside of the common context of CpG dinucleotides. Protocol: Illumina Sequencing of Bisulfite-Converted DNA Libraries (Lizardi et al. 2017b) presents a method based on the use of the Illumina platform to analyze bisulfite-converted DNA (MethylC-seq, based on the work of Lister et al. 2009 and Popp et al. 2010). This approach revealed that nearly one-quarter of all the methylation identified in embryonic stem cells occurred in a non-CG context and that this non-CG methylation disappeared upon induced differentiation of the embryonic stem cells and was restored in induced pluripotent stem cells. These findings emphasize the remarkable discovery power of this unbiased genome-wide DNA methylation analysis approach.
ADVANTAGES AND LIMITATIONS OF DIFFERENT APPROACHES FOR ANALYZING DNA METHYLATION
The choice of methods and technology platforms for DNA methylation analysis will depend on the specific biological question being addressed, the number and kind of biological samples being analyzed, the breadth and resolution of the genomic information being sought, and the budget constraints of each study.
If the amount of DNA available for analysis is limited, methods that use a DNA amplification step may be necessary. However, if the method involves the use of bisulfite modification, this step must precede the DNA amplification step so that information relevant to pattern of DNA methylation is retained. Some of the methods introduced here permit analysis of samples containing as little as 100 ng of genomic DNA, whereas other methods require 2–10 µg of sample. Remarkably, some of the whole-genome sequencing approaches, such as the high-throughput method based on the use of the Illumina platform, have been adapted to work with sample inputs as small as 150 ng (Popp et al. 2010).
If the samples to be analyzed are derived from cancer cells, the presence of deletions, translocations, regions of amplification, and other sequence rearrangements present special challenges. Affinity-based techniques must be used with caution in such cases, because the experimental results can be biased by copy number variation. On the other hand, endonuclease-based methods that measure a ratio of methylated to unmethylated DNA can be used to generate data that are less prone to distortion by locus copy number (Szpakowski et al. 2009).
Desired coverage and resolution are key considerations when choosing a method. If only a few hundred gene promoters need to be analyzed, bisulfite sequencing or MS-PCR is a reasonable choice, the former providing single-nucleotide resolution for regions of up to 800 bases, the latter offering simplicity and lower cost. For genome-wide analysis of DNA methylation, microarray-based approaches may be more economical than sequencing-based approaches. On the other hand, sequencing-based approaches offer improved resolution of methylated bases in DNA. If there is an expectation that the samples to be analyzed contain different cell types or different stages in the development of a cell lineage, sequencing-based approaches that provide information derived from molecular clones will be able to discern different methylation states at the same genomic locus. In other words, clonal sequencing will preserve “digital” information about heterogeneous patterns of methylation, thanks to the molecular sampling advantages of multiple deep-sequencing reads. For example, a rare methylation pattern occurring in one cell (perhaps a progenitor cell of interest?) out of every 200 cells in a sample will be detectable using ultradeep sequencing.
Footnotes
-
From the Molecular Cloning collection, edited by Michael R. Green and Joseph Sambrook.








