
Mapping Short Sequence Reads to a Reference Genome
Abstract
This protocol describes mapping short sequence reads to a reference genome using several programs. The example in this protocol starts with a ChIP-seq data set in FASTQ format, aligns the reads to the human genome using Bowtie, and uses some useful utilities of SAMtools and BEDTools. SAMtools and BEDTools are two collections of executables for manipulating the results of short-read aligners. By combining these tools, one can summarize and visualize alignments produced by Bowtie and perform basic analysis, such as determining the number of reads that are mapped to a certain gene. These tools can also be easily incorporated into computational pipelines of more complex analyses.
MATERIALS
Equipment
Computer equipped with Internet connection and a web browser
UNIX environment
-
The UNIX environment is required in this protocol. It is strongly recommended to work under a UNIX-based operating system, such as Ubuntu, Centos/Red Hat, or Mac OS X. If you are using a Windows operating system, install Cygwin first and install Bowtie (http://bowtie-bio.sourceforge.net/index.shtml), SAMtools (http://samtools.sourceforge.net/), and BEDTools (http://code.google.com/p/bedtools/) under Cygwin. Cygwin is free software that provides a UNIX-like environment on Windows. The Cygwin install package can be found at http://www.cygwin.com/.
METHOD
-
The output of a sequencing run is usually in FASTA or FASTQ format. FASTQ is similar to FASTA, with a header line followed by a nucleotide sequence line for each read but with an additional sequencing quality score (called Phred quality) for each base (see Introduction: Data Formats in Bioinformatics [Hung and Weng 2016]). Most of the mapping algorithms take both formats.
-
The resulting alignments can be used for peak finding in Protocol: Identifying Regions Enriched in a ChIP-seq Data Set (Peak Finding) (Hung and Weng 2017).
Creating a Data Set in the Correct Format
-
1. Obtain a ChIP-seq data set produced using an antibody against STAT1 in interferon-γ (IFN-γ)-stimulated HeLa S3 cells (GSE12782; Rozowsky et al. 2009).
-
i. Download all of the FASTQ files from the GEO website (Barrett et al. 2007; http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE12782) by following the “ftp” link for the “SPR000/SPR000703” data set (indicated by the red oval in Fig. 1).
-
ii. The linked page contains two folders, SRX003799 and SRX003800, each of which contains six compressed files. In the folder SRX003799, SRR014987.fastq.bz2 to SRR014992.fastq.bz2 are the FASTQ files for ChIP-seq against STAT1. In the folder SRX003800, SRR014993.fastq.bz2 to SRR014998.fastq.bz2 are the FASTQ files for the input (negative control that omits the antibody; see Protocol: Identifying Regions Enriched in a ChIP-seq Data Set (Peak Finding) [Hung and Weng 2017] for more information).
-
-
2. Place all of the downloaded files in a folder named “GSE12782” and decompress them using any applicable software or by executing the following UNIX command in the GSE12782 folder:
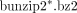
-
3. Merge all STAT1 ChIP-seq FASTQ files together.
-
i. Use the following two UNIX commands:

-
ii. Two merged files will be generated: STAT1.fastq and INPUT.fastq.
-
-
4. Obtain the human genome sequence in a format required by Bowtie by downloading and decompressing the prebuilt files on Bowtie’s website (ftp://ftp.cbcb.umd.edu/pub/data/bowtie_indexes/). The process of building such files step by step is as follows.
-
i. Go to the download page of the UCSC Genome Browser (http://hgdownload.cse.ucsc.edu/downloads.html). Click “Human” and click “Full data set” in the “Feb. 2009 (hg19, GRCh37)” section. On the bottom of the linked page, download the “hg19.2bit” file and save it in the newly created local folder “GSE12782.” This is the binary compressed version of the human genome.
-
ii. When the download is completed, another program called “twoBitToFa” is needed to convert the “hg19.2bit” file to FASTA format, which is required by Bowtie. Follow the link http://hgdownload.cse.ucsc.edu/admin/exe/, choose the appropriate computer platform (e.g., “linux.x86_64”), and click “twoBitToFa” to save this program in the local folder “GSE12782.”
-
iii. Type the command
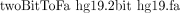
-
iv. A file named “hg19.fa” will be generated, which is the human genome split by chromosomes in FASTA format.
-
v. We need to index the human genome for fast lookup and alignment of short sequences. Type the following command:
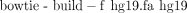
-
vi. Six index files will result: hg19.1.ebwt, hg19.2.ebwt, hg19.3.ebwt, hg19.4.ebwt, hg19.rev.1.ebwt, and hg19.rev.2.ebwt.
-
GEO web page for GSE12782. Download the FASTQ files by following the link marked with a red oval.
Aligning Reads to the Human Genome Using Bowtie
-
5. To align reads in STAT1.fastq and INPUT.fastq to the human genome by Bowtie,
-
i. Type the following two commands (see Box 1):
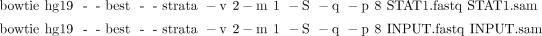
-
ii. To allow Bowtie to take advantage of the sequencing quality information in the input FASTQ file while computing alignments, change “–v” to “–n” and supply the definition of how the quality scores were computed: “–phred33-quals,” “–phred64-quals,” or “–solexa-quals.”
-
-
6. The two output alignment files STAT1.sam and INPUT.sam are in SAM format. SAM uses a rich yet compact data structure, and computer programs are required to parse out the fields of interest in a SAM file and format them into a human-friendly table. SAMtools and BEDTools are two suites of computer programs that can be used for such a purpose.
-
i. Use the following commands to convert STAT1.sam and INPUT.sam files to the binary BAM format, which is much more computationally efficient than SAM for storage and visualization:
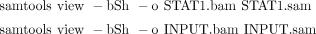 Parameter b indicates to SAMtools to output the BAM format; parameter S indicates that the input is in the SAM format; and
parameter h tells SAMtools to keep the header in the output. STAT1.bam and INPUT.bam can be uploaded to the UCSC Genome Browser
as custom tracks.
Parameter b indicates to SAMtools to output the BAM format; parameter S indicates that the input is in the SAM format; and
parameter h tells SAMtools to keep the header in the output. STAT1.bam and INPUT.bam can be uploaded to the UCSC Genome Browser
as custom tracks.
-
ii. Figure 2A shows a particular locus (chr1:175,161,500–175,163,500) of STAT1.bam. The alignments that map to the plus and minus genomic strands are colored in blue and red, respectively. Set the zoom of the alignments to the base level to see mismatches (see Box 2), revealing putative SNPs or sequencing errors.
-
-
7. These alignments can also be summarized as a histogram of the amount of reads that map to each location of the genome. The genomeCoverageBed program in BEDTools can produce such histograms and store them in the UCSC bedGraph format (http://genome.ucsc.edu/goldenPath/help/bedgraph.html). For computational efficiency, genomeCoverageBed requires the alignments to be sorted according to chromosomes, which can be accomplished by the following two steps.
-
i. Use the following commands to sort STAT1.bam and INPUT.bam according to genomic coordinates and save the result files as STAT1.sorted and INPUT.sorted:
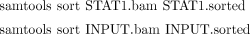
-
ii. Use the following BEDTools commands to convert the BAM files to BED files:
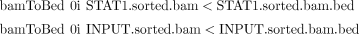 The following few lines result from a BED file:
The following few lines result from a BED file:

-
iii. genomeCoverageBed also needs to have an input file that lists the names of the chromosomes and their sizes (in base pairs), tab-delimited as follows (e.g., the human genome):

-
iv. Because the BAM files also contain such information, use the following UNIX command to create the file named human.genome:
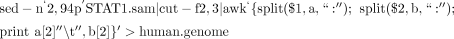
-
v. Finally, use the following commands to create four files that contain information about the amount of reads that are mapped onto each genomic position for the plus and minus genomic strands separately:

-
vi. Separate the reads that mapped to different strands of the genome by giving the parameter “-strand.” If in certain applications it is more desirable for reads on both strands to be mixed, then omit this parameter. The following is an example of several lines of one of the files:
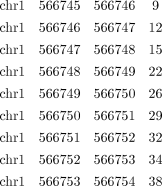
-
-
8. Directly upload the four bedGraph files to the UCSC Genome Browser as custom tracks. To decrease file size, extract a particular locus (chr1:175,161,500-175,163,500) using the following UNIX command:
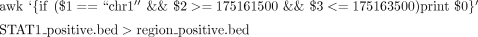 Figure 2B shows the histograms for this locus, separately for plus and minus genomic strands.
Figure 2B shows the histograms for this locus, separately for plus and minus genomic strands.
PARAMETERS IN BOWTIE
Bowtie has many parameters and it is necessary to understand the use of each parameter and choose the correct combination. See the Bowtie manual for detailed explanations of the parameters (http://bowtie-bio.sourceforge.net/manual.shtml#the–n-alignment-mode). The example in Step 5 contains the most essential components:
-
"--best --strata": for each read, report only the alignments with the fewest mismatches (see Box 2)
-
"-v 2": only report an alignment if it has two or fewer mismatches
-
"-m1": only report the alignment of a read if it maps to exactly one location on the genome (these reads are called unique mappers)
-
"-q": use FASTQ as input
-
"-S": output the alignment in SAM format
-
"-p 8": accelerate Bowtie by invoking eight searching threads at the same time on separate processors or cores. The optimal number of threads depends on available processors
CAN BOWTIE ALLOW A LARGE NUMBER OF MISMATCHES?
Unfortunately, the design of Bowtie makes it impossible to find alignments having more than three mismatches. Bowtie also does not guarantee finding all alignments with mismatches. The drawback (low sensitivity) is common in most fast short-read alignment programs, because they are derived from exact string-matching algorithms for speed gains at the cost of sensitivity (see Box 3). Allowing more mismatches would significantly reduce the speed. If sensitivity is a concern in an application, one might want to realign the unmappable region using BLAST to recover additional alignments.
OTHER FAST SHORT-READ ALIGNERS
The fastest algorithms for aligning short reads to a reference genome include Bowtie, BWA, and SOAP2. They adapt similar BW transforms to compress the suffix/prefix tree storing a genome. The differences among the algorithms are their strategy toward mismatches, gaps, and paired-end reads. A comparison of these tools can be found in Li and Durbin (2009) and Li and Homer (2010). Simulation has shown that SOAP2 was the fastest and BWA the most accurate. Note that SOAP2 needs about twice the memory of BWA and Bowtie. In general, speed comes with the cost of accuracy and these aligners are all highly competitive.
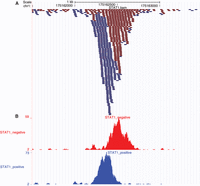
Visualizing the Bowtie alignments on the UCSC Genome Browser. (A) The uploaded STAT1.bam file. By setting the zoom to the base level, mismatches in each read will become visible. (B) Two custom tracks uploaded to the UCSC Genome Browser. Leftmost labels indicate the names of the tracks.
DISCUSSION
Most studies of small RNA deep sequencing data extract reads that map perfectly to the genome (sometimes allowing a few mismatches to account for sequencing errors) and discard the reads that do not map. However, because SNPs, transposons, repeats, and structure variants are common in most organisms, it is likely to see many reads that do not align to the reference genome; these could in fact be interesting biological results. For example, flies that lack hen1, which is a methyltransferase that adds a 2′-O-methyl group to the 3′ end of endo-siRNAs and Piwi-interacting RNAs (piRNAs), show extensive trimming and tailing of endo-siRNAs and piRNAs. These trimmed and tailed sequences cannot be mapped to the reference genome by standard short-read mapping algorithms. A suffix-tree-based algorithm was designed to discover these reads and all of the genomic locations of their unmodified precursors (Ameres et al. 2010, 2011).
Footnotes
-
From the Molecular Cloning collection, edited by Michael R. Green and Joseph Sambrook.








