
Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) Technology in Fission Yeast
- Boris Maček1,7,
- Alejandro Carpy1,
- André Koch2,4,
- Claudia C. Bicho3,5,
- Weronika E. Borek3,
- Silke Hauf2,6 and
- Kenneth E. Sawin3,7
- 1Proteome Center Tuebingen, Interfaculty Institute for Cell Biology, University of Tuebingen, 72076 Tuebingen, Germany
- 2Friedrich Miescher Laboratory of the Max Planck Society, Tuebingen, 72076, Germany
- 3Wellcome Trust Centre for Cell Biology, School of Biological Sciences, University of Edinburgh, Edinburgh EH9 3BF, United Kingdom
- ↵7Correspondence: boris.macek{at}uni-tuebingen.de; ken.sawin{at}ed.ac.uk
Abstract
Shotgun proteomics combined with stable isotope labeling by amino acids in cell culture (SILAC) is a powerful approach to quantify proteins and posttranslational modifications across the entire proteome. SILAC technology in Schizosaccharomyces pombe must cope with the “arginine conversion problem,” in which isotope-labeled arginine is converted to other amino acids. This can be circumvented by either using stable isotope-marked lysine only (as opposed to the more standard lysine/arginine double labeling) or using yeast genetics to create strains that only very inefficiently convert arginine. Both strategies have been used successfully in large-scale (phospho)proteomics projects in S. pombe. Here we introduce methods for performing a typical SILAC-based experiment in fission yeast, including generation of SILAC-compatible strains, sample preparation, and measurement by mass spectrometry.
OVERVIEW OF SILAC
Shotgun proteomics has emerged as a state-of-the-art technology to globally identify and quantify proteins and posttranslational modifications using liquid chromatography combined with mass spectrometry (LC–MS). This technique is semiquantitative and must be combined with other chemical, biological, or bioinformatic techniques to yield quantitative data (Ong et al. 2002; Thompson et al. 2003; Boersema et al. 2009; Luber et al. 2010). One of the major approaches in quantitative proteomics is metabolic labeling of cells with stable, nonradioactive “heavy” isotopes (e.g., 2H, 13C, and 15N). This can be achieved by labeling the whole proteome with 15N (Washburn et al. 2002) or by stable isotope labeling by amino acids in cell culture (SILAC). SILAC is an elegant method for relative quantification of peptides and proteins from different samples (Ong et al. 2002; Ong and Mann 2006). It involves metabolic labeling of cells with exogenous amino acids containing defined numbers of stable isotopes, thereby introducing a defined isotope label into newly synthesized cellular proteins. A separate cell culture is grown in the presence of the corresponding natural “light” amino acids. Heavy and light cultures can either correspond to two different treatments or growth conditions of the same strain or to two different strains altogether. Heavy and light cell lysates are mixed in equal amounts according to cell count or protein content. The combined protein extract is digested with an appropriate protease, and the resulting peptide mixture is measured in a mass spectrometer using nanoflow liquid chromatography–tandem mass spectrometry (nano LC–MS/MS) (Fig. 1). In a SILAC MS measurement, peptide ions are detected as doublets, and the total signal ratio between the members of a doublet corresponds to the relative difference in the amount of the relevant protein or peptide (e.g., for a posttranslational modification) between the two analyzed states. Because most of the amino acids used for labeling allow for synthesis of two different heavy forms, SILAC can be extended to three samples, often called “heavy,” “medium-heavy,” and “light”; in this case, peptide ions are detected as triplets.
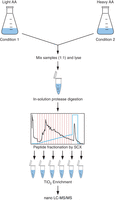
SILAC-based phosphoproteomics workflow using light and heavy amino acids (AA); see Protocol: Construction, Growth, and Harvesting of Fission Yeast Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) Strains (Koch et al. 2016) and Protocol: Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)-Based Quantitative Proteomics and Phosphoproteomics in Fission Yeast (Carpy et al. 2016). SCX, Strong cation exchange.
LABELING PROTEINS: CHALLENGES IN FISSION YEAST
An ideal SILAC growth medium contains both heavy arginine and heavy lysine. This is because trypsin—the standard protease for digestion of proteins because it creates peptide sizes suitable for MS—cleaves polypeptide chains after arginine and lysine residues. Labeling with heavy arginine and lysine ensures that all peptides contain at least one heavy amino acid and therefore are quantifiable. (Because of incomplete digestion, some peptides may contain more than one heavy amino acid.) Because wild-type Schizosaccharomyces pombe can synthesize all amino acids, labeling cells with heavy amino acids requires the use of auxotrophic strains. Labeling fission yeast lysine-auxotrophic cells with heavy lysine is straightforward and does not present any problems (Koch et al. 2011; Wang et al. 2012). However, arginine is catabolized and converted into other amino acids, including but not limited to proline. As a result, heavy label can be incorporated into proteins at amino acid residues other than arginine, leading to the appearance of additional peaks in MS and MS/MS spectra of a given peptide. This so-called “arginine conversion problem” can seriously confound peptide identification and quantification. In addition, certain auxotrophs in fission yeast, including lysine/arginine double auxotrophs, can show impaired growth even in the presence of the required amino acids. Therefore, SILAC experiments in fission yeast face two challenges: (1) avoiding catabolic conversion of labeled arginine to other amino acids; and (2) obtaining robust growth of auxotrophic strains in labeling experiments.
In mammalian cells and budding yeast, arginine conversion is minor, and several approaches have been developed to minimize the problem, including titration of arginine and proline concentrations and computational methods (Gruhler et al. 2005; Blagoev and Mann 2006; Ong and Mann 2006; Bendall et al. 2008; Graumann et al. 2008; Mousson et al. 2008; Park et al. 2009; Prokhorova et al. 2009). In fission yeast, arginine conversion is much more extensive (Bicho et al. 2010) and thus must be dealt with more decisively. Two general approaches are possible, and both have been used successfully (Bicho et al. 2010; Koch et al. 2011). The first approach (Koch et al. 2011) avoids heavy arginine altogether. Instead, cells are labeled only with heavy lysine, and, in place of trypsin, proteolytic digestion into peptides is performed using lysyl endopeptidase Lys-C, which cleaves after lysine residues. A potential disadvantage of this approach is that peptides produced by Lys-C digestion are significantly longer than those produced by trypsin. This can affect the “flyability” of peptides and thus identification in the mass spectrometer. The second approach (Bicho et al. 2010) takes advantage of yeast genetics and deletes genes that encode enzymes involved in arginine catabolism, thereby preventing or at least decreasing arginine conversion. Deletion of the predicted ornithine transaminase gene car2+ or simultaneous double deletion of the redundant predicted arginase genes car1+ and aru1+ has been shown to decrease conversion to acceptably low levels (Bicho et al. 2010). Because constructing single-deletion strains is easier, deletion of the car2+ gene has generally been the preferred method.
PROTOCOLS FOR FISSION YEAST
We describe construction of fission yeast strains suitable for SILAC MS, as well as conditions for robust growth, harvesting, and processing of cells (Bicho et al. 2010; Koch et al. 2011) in Protocol: Construction, Growth, and Harvesting of Fission Yeast Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC) Strains (Koch et al. 2016). We also provide a method for enrichment and analysis of protein Ser/Thr/Tyr phosphorylation in fission yeast using strong cation exchange (SCX) and TiO2 chromatography, followed by nano LC–MS/MS, in Protocol: Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)-Based Quantitative Proteomics and Phosphoproteomics in Fission Yeast (Carpy et al. 2016). This strategy has been used in several large-scale phosphoproteomics studies in eukaryotes (Olsen et al. 2006, 2009) and prokaryotes (Maček et al. 2007, 2008). The protocol follows methods outlined in Maček et al. (2009) (Fig. 1) and presents a revised version of a previously published protocol (Olsen and Maček 2009). Parts of this protocol can be applied to enrichment and analysis of other posttranslational modifications. For example, SCX can be used to enrich for amino-terminal acetylation (Helbig et al. 2010), and TiO2 can be used to enrich for sugars containing negatively charged residues, such as sialic acid (Larsen et al. 2007). We focus on enrichment of phosphopeptides and note how the protocol can be adapted for proteome analysis. Other methods can be used if deeper proteome coverage is required (Shevchenko et al. 1996; Hubner et al. 2008).
ACKNOWLEDGMENTS
Our work is supported by funding from the Juniorprofessoren-Programm of the BW-Stiftung and SFB 766 of the German Research Council (DFG) to B.M. and by a Wellcome Trust grant (094517) to K.E.S., A.K., and S.H. received funding from SFB 446 of the German Research Council (DFG) and the Max Planck Society. W.E.B. was supported by a Cancer Research UK PhD Studentship (C20060/A10789). The Wellcome Trust Centre for Cell Biology is supported by core funding from the Wellcome Trust (092076).
Footnotes
-
From the Fission Yeast collection, edited by Iain M. Hagan, Antony M. Carr, Agnes Grallert, and Paul Nurse.








