
3C-Based Chromatin Interaction Analyses
Abstract
This introduction presents a molecular approach that uses formaldehyde cross-linking to investigate genome structure and function—chromosome conformation capture (3C). This approach allows us to determine the spatial proximity of distant functional genomic sites (by looping). 3C-based techniques to interrogate chromosome folding and long-range interactions between genomic sequences in vivo are detailed.
3C-BASED CHROMATIN INTERACTION ANALYSES
Chromosomes are folded in three dimensions, and their spatial arrangements plays roles in gene regulation, chromosome segregation, genome stability, and possibly other aspects of chromatin metabolism. Specifically, long-range interactions can occur between widely spaced genomic elements, for example, between genes and their distant regulatory elements, which can be located up to several megabases away. These interactions result in the formation of chromatin loops.
Several related methods have been developed, initially pioneered by Dekker et al. (2002), that use formaldehyde cross-linking to trap, detect, and quantify physical interactions between genomic loci, both along and between chromosomes. The central component of all these methods is the 3C assay (Dekker et al. 2002). 3C allows the determination of the frequency of interaction of any pair of small loci (up to a few kilobases) throughout a genome inside intact cells. 3C is widely used to detect looping interactions between genomic elements, for example, enhancers and promoters (Dekker 2003, 2008; Simonis et al. 2007). We describe detailed associated protocols for performing 3C, as well as several variations of 3C, including two high-throughput adaptations that allow large-scale detection of chromatin interactions throughout genomes.
Outline of the 3C Technique
3C is based on formaldehyde cross-linking of intact cells that covalently links physically touching genomic loci by cross-linking proteins to DNA and proteins to proteins. Next, 3C uses a series of molecular manipulations to determine the identity of the interacting loci. First, cross-linked chromatin is solubilized using a combination of detergents, mechanical lysis of cells, and brief incubation at 65°C. Then chromatin is digested with a restriction enzyme, and, finally, DNA is ligated under dilute conditions that favor intramolecular ligation of cross-linked chromatin segments. Cross-links are then reversed and the ligation mixture is purified. 3C yields a genome-wide ligation product library in which each ligation product corresponds to a specific interaction between the two corresponding loci. The frequency with which a specific 3C ligation product occurs in the library is a measure of the frequency with which the loci are sufficiently close in space to be cross-linked.
The next step is quantitative detection of 3C ligation products. This step is what distinguishes the various 3C-based methods from one another. Conventional 3C uses polymerase chain reaction (PCR) with specific primers to detect 3C ligation products one at a time. The PCR primers are designed to anneal 100–150 bp upstream of and downstream from the newly formed restriction site of the ligation product (Fig. 1). PCR products are displayed on a gel and then quantified using a gel quantification system. Alternatively, real-time PCR with TaqMan primers is used to quantify the abundance of a specific 3C ligation product (Hagege et al. 2007). Both methods yield very similar results. The amount of PCR product that is formed is a measure of the frequency with which the loci interact.

Schematic outline of 3C-based technologies. (A) Cross-linked chromatin segments (light blue and dark blue lines) connected by a protein complex (gray circle) are digested with a restriction enzyme (sites indicated by black bars) and then ligated. In the standard 3C protocol, DNA cross-links are then reversed and DNA is purified. In the ChIP-loop protocol, ligated chromatin is first immunoprecipitated with a specific antibody against a protein of interest, and then cross-links are reversed and DNA is purified. Both protocols yield a 3C ligation product library that is composed of both linear and circular DNA molecules. (B) Three methods for analysis of the 3C ligation product library. In the conventional 3C protocol, ligation products are detected by PCR with primers that amplify the ligation junction (primers are indicated with arrows; the arrowhead is the 3′ end of the primer). The amplified DNA fragments are detected and quantified on an agarose gel. In the 4C protocol, inverse PCR is used to amplify all fragments ligated to a single fragment of interest. The resulting amplified DNA is analyzed on a microarray or by deep (high-throughput) sequencing. In the 5C protocol, sets of ligation products are detected by multiplex ligation-mediated amplification. 5C primers are designed to anneal adjacent to each other across the ligation junction; arrowheads indicate the 3′ ends. Note the presence of universal tails (gray line/arrow) on the 5C primers that do not anneal to the 3C ligation product. See Figure 2 of details of primer design. The resulting amplified DNA is analyzed on a microarray or by deep sequencing.
Control Libraries
PCR detection of 3C ligation products relies on the use of specific primer pairs to detect individual ligation products. These primer pairs may differ in their annealing temperature and amplification efficiency. To correct for these differences, a control ligation library can be constructed that contains all possible ligation products in equal abundance. Interaction frequencies are then determined as follows: PCR is used to amplify a specific ligation product in the 3C library as well as in the control library. The ratio of the amount of PCR product obtained with the 3C library to the amount of PCR product obtained with the control library is then calculated. This ratio is a measure for the interaction frequency of the two loci and is normalized for any differences in primer efficiency or amplification efficiency of the target DNA.
Control libraries can be generated by randomly ligating restriction enzyme-digested purified genomic DNA. This method is only suitable for small genomes, including those of bacteria and yeast. For larger genomes (e.g., those of human and mouse), this method yields libraries that are too complex to allow accurate detection of individual ligation products. For larger genomes, a control library should be made that represents only the section of the genome in which the researcher is interested. Most 3C studies are limited to analysis of only a few hundred kilobases. In that case, combining DNA of one or more BAC clones that together represent the genomic region of interest can generate control libraries.
3C Applications
Conventional 3C with PCR detection of 3C ligation products is used for targeted analysis of relatively small (up to a few hundred kilobases) genomic regions to identify looping interactions between candidate genomic elements. Because interactions are detected in a one-by-one fashion, the number of chromatin interactions that can be tested is usually limited to up to a few dozen. In addition, control libraries are always used to correct for differences in PCR primer and amplicon amplification efficiencies.
Outline of the 4C Technique
Several methods have been developed to detect 3C ligation products in a 3C library in a high-throughput setting. These methods use the standard 3C method to generate libraries of 3C ligation products but then use various assays for large-scale detection of sets of ligation products. The 4C technique—3C-on chip, or circular chromosome conformation capture (Simonis et al. 2006; Zhao et al. 2006; Gondor et al. 2008)—uses inverse PCR to amplify all loci that have become ligated to a single locus of interest during the 3C assay. 3C typically yields both linear and circular ligation products (Fig. 1). To generate smaller circular molecules, purified 3C ligation libraries can be digested with a frequently cutting restriction enzyme and then recircularized by intramolecular ligation (Simonis et al. 2006). Then inverse PCR primers are designed on each end of the locus of interest (the “bait” fragment) so that the intervening unknown ligation partner can be amplified by PCR. The sequence of the amplified DNA is then determined using genome-wide microarrays or by high-throughput sequencing. 4C yields genome-wide interaction profiles that can identify genomic regions that are frequently close in space to the one locus of interest.
When 4C is performed on cells with small genomes, such as bacteria and yeast, a control ligation product can be generated as described above for the 3C method. 4C can be performed on this control library using the same set of inverse PCR primers, and the resulting interaction profile serves as a control that can be used to normalize for differences in amplification efficiency exactly as described for 3C above. When 4C is performed with cells that contain larger genomes, such as human and mouse cells, a representative control library cannot easily be constructed because of the extremely high complexity of the resulting ligation mixture, as discussed above for 3C.
4C Applications
4C is used to identify loci throughout the genome that can interact with a specific genomic element of interest. For instance, one can identify all genomic regions that interact with a gene or regulatory element of interest.
Outline of the 5C Technology
The 5C technology—3C-carbon copy (Dostie et al. 2006; Dostie and Dekker 2007)—also starts with the conventional 3C protocol, but it then interrogates the 3C library using highly multiplexed ligation-mediated amplification with pools of locus-specific primers, followed by quantification of ligated primer pairs using microarrays or high-throughput sequencing (Fig. 1). The hallmark of 5C is the use of large pools of up to thousands of primers in a single reaction to allow detection of large numbers of 3C ligation products in parallel.
5C Primer Design
Restriction fragments of interest are selected throughout the genome, and a 5C primer is designed for each of them. 5C uses two types of primers—forward primers and reverse primers (Fig. 2). Either a forward or a reverse primer is designed for each restriction fragment. Typically, 5C primers are designed so that head-to-head 3C ligation products are detected. Forward primers are designed to anneal to the bottom strand (as displayed in the linear genome assembly) precisely at the 3′ end of a restriction fragment, including half of the restriction site. Reverse primers are designed to anneal to the top strand (as displayed in the linear genome assembly) precisely at the 3′ end of a restriction fragment, including half of the restriction site. Thus, when aligned along the assembled genome, forward and reverse primers anneal to opposite strands (Fig. 2). However, a combination of forward and reverse primers will anneal to the same strand directly adjacent to each other across the new restriction site of a specific head-to-head 3C ligation product. In that configuration, pairs of primers can be ligated using nick-specific ligases such as Taq ligase. This 5C primer design only detects interactions between restriction fragments that are recognized by a forward primer and fragments that are recognized by a reverse primer. Finally, forward and reverse primers carry universal tails (at the 5′ end for forward primers and at the 3′ end of reverse primers). These universal tails allow PCR amplification of only ligated pairs of forward and reverse 5C primers using a single pair of PCR primers.
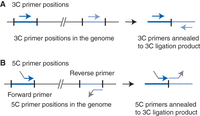
Primer positions for 3C and 5C. (A) Primers used for PCR detection of 3C ligation product are designed so that they anneal to the same strand of genomic DNA and are able to prime amplification of a head-to-head 3C ligation product. (B) Primers used for 5C detection are designed so that they anneal to the opposite strands of genomic DNA and are able to detect a head-to-head 3C ligation product. Arrowheads on primers indicate the 3′ ends. The nonannealing gray sections of the 5C primers represent the universal tails (see the main text). Forward primers have a 5′ universal tail, whereas reverse primers carry a universal tail at their 3′ ends.
When 5C is performed with cells that contain small genomes, a control ligation product library can be generated as described above for the 3C method. 5C is then performed on this library using the same pool of 5C primers. The resulting library of ligated primers pairs serves as a control that can be used to normalize for differences in primer annealing and amplification, as described for 3C. When 5C is performed with cells that contain larger genomes, a representative control library can only be constructed using a set of BAC clones (as described above for 3C) when a relatively small segment of the genome is analyzed (up to several megabases). When loci are spread over larger regions, construction of a control is not feasible. It is important to note that 5C is much less sensitive to differences in primer efficiency than regular PCR, and thus the need for a control library is reduced when compared with 3C. This is because 5C primers are typically designed to be of equal length (thus amplicons of different combinations of forward and reverse primers are identical in length) and to display the same annealing temperature so that differences in primer annealing and amplification are typically small, reducing the need to control for these differences. In addition, the fact that amplification of ligated 5C primer pairs is achieved with a single universal pair of PCR primers further reduces the differences in amplification efficiency of different 5C primer pairs.
5C Applications
5C allows analysis of all interactions between two sets of genomic loci—one set that is recognized by forward primers and one set that is recognized by reverse primers. 5C can be performed at very high levels of multiplexing using thousands of 5C primers (Dostie et al. 2006; Lajoie et al. 2009). For instance, by combining 1000 forward and 1000 reverse 5C primers, 5C will interrogate all 1 million pairwise chromatin interactions between the corresponding 2000 restriction fragments in a single assay.
Two types of applications can be distinguished. First, one type of primers (e.g., reverse primers) is designed for one set of loci of interest (e.g., gene promoters), and the second set (forward primers) is designed for a separate set of loci of interest (e.g., gene regulatory elements). A 5C analysis with this set of primers will allow the parallel detection of any looping interactions between these promoters and gene regulatory elements.
A second application involves generating dense interaction matrices throughout genomic regions. “Dense interaction matrices” are data sets that represent interactions between many closely spaced restriction fragments. These data sets can be used to identify novel, long-range, looping interactions between pairs of loci that were not previously known to be interacting, or known to contain functional elements. Dense interaction matrices can also be used to obtain information about the overall three-dimensional (3D) folding of a genomic region, as has been done for yeast chromosome III (Dekker et al. 2002). Dense interaction matrices are generated when forward and reverse 5C primers are designed in an alternating fashion for consecutive restriction fragments throughout the region of interest. 5C will then detect interactions between all odd-numbered and all even-numbered restriction fragments, numbering the fragments according to their genomic order. 5C can be performed with up to several thousands of 5C primers, and the genomic region that can be analyzed in this manner can therefore be up to tens of megabases in size.
The Chip-Loop Technique
The Chip-loop technique (Horike et al. 2005; Tiwari et al. 2008) combines 3C with chromatin immunoprecipitation (ChIP) (Fig. 1). For information about ChIP see Introduction: Chromatin Immunoprecipitation (ChIP) Analysis of Protein-DNA Interactions (Kim and Dekker 2018a). The Chip-loop protocol starts by cross-linking cells with formaldehyde. After solubilization and restriction digestion, chromatin is immunoprecipitated with an antibody that recognizes a protein of interest. As a result, chromatin segments that are bound by this protein are precipitated selectively. When these segments are interacting with other genomic loci, these regions are also pulled down. The steps that follow are again similar to 3C again and include ligation of cross-linked chromatin segments, reversal of cross-links, and purification of the ligation mixture.
Analysis of the Chip-loop ligation mixture is performed exactly as described for 3C ligation libraries. PCR can be used to detect individual ligation products one at a time, 4C can be used to detect all loci that were ligated to a specific locus of interest, or 5C can be used to detect interactions between two large sets of elements.
Chip-Loop Applications
This method is used to determine whether proteins of interest are present when two loci are engaged in a specific looping interaction.
Choosing Which 3C-Based Assay to Use
Several considerations determine which 3C-based method is required to address a specific research question. All 3C-based methods start with the basic 3C protocol but then differ in the method for detection of 3C ligation products. Table 1 outlines the specific features of the various 3C-based methods and can serve as a guideline for deciding which detection method is needed for a specific research project.
Overview of the features of 3C-based methods
Overview of Protocols for 3C-Based Methods
All 3C-based methods start with cross-linking cells with formaldehyde as described in Protocol: Formaldehyde Cross-Linking (Kim and Dekker 2018b). 3C, 4C, and 5C then proceed with Protocol: Generation of 3C Libraries from Cross-Linked Cells (Kim and Dekker 2018c), whereas the Chip-loop method continues with Protocol: Generation of ChIP-Loop Libraries (Kim and Dekker 2018d). Protocol: Generation of Control Ligation Product Libraries for 3C Analyses (Kim and Dekker 2018e) describes how random ligation product libraries can be generated that can serve as a normalization control for 3C, 4C, 5C, and Chip-loop assays. The following three protocols then describe various methods to detect ligation products in 3C libraries or Chip-loop libraries. Protocol: Polymerase Chain Reaction (PCR) Detection of 3C Ligation Products Present in 3C, ChIP-Loop, and Control Libraries: Library Titration and Interaction Frequency Analysis (Kim and Dekker 2018f) describes semiquantitative PCR to analyze 3C, Chip-loop, and control libraries. Protocol: 4C Analysis of 3C, ChIP-Loop, and Control Libraries (Kim and Dekker 2018g) describes the 4C method for detecting genome-wide interaction profiles of candidate loci. Protocol: 5C Analysis of 3C, ChIP-Loop, and Control Libraries (Kim and Dekker 2018h) describes the 5C method for detecting chromatin interaction networks and dense matrices of chromatin interactions.
Expected Results and Interpretation of 3C-Based Data
Because 3C-based approaches yield relative interaction frequencies between pairs of loci, one should expect to detect frequent interactions between loci that are located near each other in the genome. Directly adjacent restriction fragments show the highest interaction frequency, and the interaction frequency declines rapidly for fragments separated by increasing genomic distances. In general, interactions become difficult to detect using semiquantitative PCR for sites separated by more than several hundreds of kilobases. 4C and 5C are more sensitive and allow detection of interactions between loci separated by tens to hundreds of megabases, and also interactions located on different chromosomes. In the absence of any specific higher-order organization of chromosomes, one would expect to detect only general background interactions that are inversely related to genomic distance.
Chip-loop experiments should also detect frequent interactions between loci located near each other along the linear genome sequence. However, owing to the ChIP enrichment step, a much lower background level of interactions and more pronounced peaks of interaction can be expected, although this will depend on the quality of the antibody.
Higher-order chromatin structures are identified by analyzing interaction data and identifying interaction frequencies that are significantly higher (or lower) than expected. The formation of specific chromatin loops between two genomic loci (e.g., a promoter and a distant enhancer) can be detected because their interaction frequency is significantly higher than expected for two loci separated by a similar genomic distance. A typical 3C-based experiment identifies a set of interaction frequencies throughout the region of interest. Chromatin loops appear as peaks in the interaction profile (discussed in detail in Dekker 2006).
All 3C-based methods report the population average interaction frequencies of loci, and this has to be taken into account when interpreting the output data and when comparing the results with data obtained by microscopy in single-cell observations. For instance, a 4C experiment can identify dozens to hundreds of regions throughout the genome that can be in close spatial proximity to the bait fragment. However, single-cell microscopy often shows that in a given cell, the bait only interacts with one or a few of these loci. One possible explanation for this apparent difference is that the spatial organization of chromosomes is extremely variable and fluctuates dramatically between cells in the population.
Finally, to be able to compare interaction data obtained in different experiments, it is critical to include a set of internal controls in each analysis. The controls are typically interactions that are not expected to be different in frequency between different experiments, conditions, or cell types. We recommend using a set of at least 10–20 interactions, separated by up to 50 kb, in a region of the genome that is expected to be identical between experiments. We have successfully used the gene desert (a region devoid of genes) on human chromosome 16 (Dostie et al. 2006) or a region with housekeeping genes (Gheldof et al. 2006). This set of interactions is then measured in all experiments, and the values obtained for them are used to calculate a normalization factor so that data from different experiments can be compared quantitatively. This normalization factor is calculated by determining for each of the interactions the log ratio of the signal obtained in one experiment over the signal obtained in the second experiment. Then the average of the entire set of log ratios is calculated. This is the normalization factor.
WHAT IS CAPTURED BY 3C-BASED ASSAYS?
3C-based methods allow the determination of the relative frequency with which pairs of loci are near each other in three dimensions inside nuclei in the cell population. As with any other interaction detection assay, these experiments do not directly reveal the molecular nature or the biological meaning of these spatial associations. Further experimentation using complementary functional analyses is required to link long-range chromosomal interactions to specific functions, such as gene regulation, and to identify the molecular machineries driving these phenomena.
3C-based assays are widely used to study the spatial organization of chromosomes, and from the many detailed studies that have been performed, it appears that the following types of chromosomal interactions can be captured (Simonis et al. 2007; Dekker 2008). First, direct physical associations between gene promoters and their gene regulatory elements, such as enhancers and insulators, are readily detected. These interactions are frequent, occur at precise cis-elements (several hundreds of base pairs in size), are dependent on transcription factor binding to the two interacting loci, and often correlate with the expression of the gene. Second, associations between groups of active genes can be detected across the genome. These interactions are less frequent and occur over large genomic regions (up to hundreds of kilobases). These interactions may reflect the spatial clustering of genes at sites in the nucleus enriched in splicing factors (speckles) (Brown et al. 2008) or RNA polymerase (sometimes referred to as “transcription factories”) (Osborne et al. 2004). Finally, and related to the interactions between groups of active genes, long-range interactions can be detected that reflect the general organization of the chromosomes inside the nucleus. These include frequent associations between pairs of complete chromosomes that reflect their preferential subnuclear positions and interactions that reflect the spatial compartmentalization of the entire genome in active and inactive neighborhoods (Simonis et al. 2006; Lieberman-Aiden et al. 2009). When analyzing and interpreting any 3C-based data, it is important to consider all these different types of interactions and to perform additional experiments to determine which class of interactions is being detected.
ACKNOWLEDGMENTS
We thank Yeun Hee Kim, Celeste Greer, and members of the Kim and Dekker laboratories for comments. The Kim laboratory has been supported by grants from the National Institutes of Health (NIH), Rita Allen Foundation, Sidney Kimmel Cancer Research Foundation, Yale Cancer Center, and Alexander and Margaret Stewart Trust. The Dekker laboratory has been supported by grants from NIH (HG003143, HG004592) and a W.M. Keck Foundation Distinguished Young Scholar in Medical Research Award.
Footnotes
-
From the Molecular Cloning collection, edited by Michael R. Green and Joseph Sambrook.








