
How to Win the Battle with RNase
Abstract
Because ribose residues carry hydroxyl groups in both the 2′ and 3′ positions, RNA is chemically much more reactive than DNA and is easy prey to cleavage by contaminating RNases—enzymes with various specificities that share the property of hydrolyzing diester bonds linking phosphate and ribose residues. Because RNases are released from cells following lysis and are present on the skin, constant vigilance is required to prevent contamination of glassware and bench tops and the creation of aerosols carrying RNase. The problem is compounded because there is no simple method to inactivate RNases. Because of the presence of intrachain disulfide bonds, many RNases are resistant to prolonged boiling and mild denaturants and are able to refold quickly when denatured. Unlike many DNases, RNases do not require divalent cations for activity and thus cannot be easily inactivated by the inclusion of ethylenediaminetetraacetic acid (EDTA) or other metal ion chelators in buffer solutions. The best way to prevent problems with RNase is to avoid contamination in the first place.
SOURCES OF RNase CONTAMINATION
Many an experiment has been needlessly ruined by contamination with RNase. However, problems with exogenous RNase can be entirely avoided by vigilant use of prophylactic measures and the prudent application of common sense. In our experience, contamination with exogenous RNase most frequently arises from two sources.
-
1. Contaminated buffers. By careless use of aseptic technique, buffers have become contaminated with bacteria or other microorganisms. The growth of these microorganisms is not usually visible to the naked eye and need not be florid to cause problems. Because RNase cannot be removed by autoclaving, solutions that are contaminated, or are suspected of being so, must be discarded.
-
2. Automatic pipetting devices. There is simply no point in using disposable pipette tips that are free of RNase if the automatic pipettor has been used previously to dispense solutions containing RNase (e.g., during processing of small-scale plasmid preparations or, even worse, in ribonuclease protection assays). If the barrel or the metal ejector of the automatic pipettor comes in contact with the sides of tubes, it becomes a very efficient vector for the dissemination of RNase.
TIPS FOR AVOIDING RNase CONTAMINATION
A mantric belief in the power of rubber gloves to ward off problems with RNase has taken root in many laboratories. In truth, however, snapping on a pair or two of rubber gloves is about as useful as carrying a rabbit's foot. First, the hair or beards of investigators are more likely to be the culprits than the hands, and more significantly, gloves can only provide protection until they touch a surface that has been in contact with skin. To be of any use at all, gloves must be changed every time a piece of apparatus is touched, a refrigerator opened, an ice-bucket filled, an entry written in a laboratory notebook, or a reagent measured. This is neither wise nor practicable. Wear gloves, but do not believe that they offer protection against RNase. More sensible measures include the following.
-
Keep a special set of automatic pipettors for use when handling RNA.
-
Set aside items of glassware, batches of plasticware, and buffers that are to be used only for experiments with RNA.
-
Store solution/buffers in small aliquots, and discard each aliquot after use. Avoid materials or stock solutions that have been used for any other purposes in the laboratory.
-
Set aside special electrophoresis devices for use in the separation of RNA. Clean these devices with detergent solution, rinse in H2O, dry with ethanol, and then fill with a 3% solution of H2O2. After 10 min at room temperature, rinse the electrophoresis tank thoroughly with H2O that has been treated with DEPC (please see the section Diethylpyrocarbonate).
-
Prepare all solutions and buffers with RNase-free glassware, DEPC-treated water, and chemicals reserved for work with RNA that are handled with disposable spatulas or dispensed by tapping the bottle rather than using a spatula. Wherever possible, treat solutions with 0.1% DEPC for at least 1 h at 37°C, and then autoclave for 15 min at 15 psi (1.05 kg/cm2) on liquid cycle.
-
Autoclaving glassware and plasticware may not be sufficient to inactivate RNase. Bake glassware for 4 h at 300°C. Treat plasticware either with DEPC or commercially available products that inactivate RNase upon contact (e.g., RNaseZap from Ambion).
-
Use disposable tips and microcentrifuge tubes certified by a reputable manufacturer to be free of RNase. To reduce the chances of contamination, it is best to use sterile forceps when transferring these small items from their original packages to laboratory racks.
-
Use inhibitors to suppress RNases during the isolation of RNA (please see the section Inhibitors of RNases).
INHIBITORS OF RNases
RNases are robust and powerful enzymes that seriously threaten the integrity of RNA at all stages of its isolation and characterization. Three types of inhibitors are commonly used to keep the activity of RNases in check.
-
DEPC is a highly reactive alkylating agent that is used to inactivate RNases in buffers and on glassware. Because DEPC indiscriminately modifies proteins and RNA, it cannot be used during isolation and purification of RNA and is incompatible with some buffers (e.g., Tris). For further details, please see the section Diethylpyrocarbonate.
-
Vanadyl ribonucleoside complexes are transition-state analogs that bind to the active sites of many RNases and inhibit their catalytic activity almost completely (Berger and Birkenmeier 1979). Because vanadyl ribonucleases do not covalently modify RNases, they must be used at all stages of RNA extraction and purification. However, because these complexes inhibit RNA polymerases and in vitro translation, they must be removed from the final preparation of RNA by multiple extractions with phenol containing 0.1% hydroxyquinoline. Vanadyl ribonucleoside complexes are available from several commercial suppliers.
-
Protein Inhibitors of RNases. Many RNases bind very tightly, albeit noncovalently, to ∼50-kDa proteins found in the cytoplasm of virtually all mammalian tissues and can be isolated in abundance from placenta (Blackburn et al. 1977). In vivo, these proteins function as inhibitors of proteins belonging to the pancreatic RNase superfamily, notably angiogenin, a blood-vessel-inducing and eosinophyl-derived neurotoxin. The affinities of these protein inhibitors for their targets are among the highest on record (1–70 fM) (Lee et al. 1989; for review, see Lee and Vallee 1993).
The archetypal RNase inhibitor is a horseshoe-shaped molecule, containing seven alternating leucine-rich repeats, 28 and 29 residues in length. The inhibitor also contains a large number of cysteinyl residues, all in the reduced form. The interface between ribonuclease and the inhibitor is unusually large and encompasses residues located in multiple domains of both proteins. However, the energetically important contacts involve only the carboxy-terminal segment of the inhibitor and the catalytic center of ribonuclease, including a crucial lysine residue (Kobe and Deisenhofer 1993, 1995, 1996; Papageorgiou et al. 1997; for review, see Hofsteenge 1994).
Protein inhibitors of RNase derived from several sources are sold by many manufacturers under various trade names (e.g., RNasin, Promega; SUPERase•In, Ambion). Although these vary in their requirement for sulfhydryl reagents, all of them display a broad spectrum of inhibitory activities against RNases but do not inhibit other nucleases or polymerases or in vitro translation systems (e.g., see Murphy et al. 1995).
Because the inhibitors do not form covalent complexes with RNase, they cannot be used in the presence of denaturants such as SDS and guanidine, which are commonly used to lyse mammalian cells in the initial stages of extraction of RNA. However, the inhibitors can be included at all stages during subsequent purification of RNA. Inhibitors must be replenished several times during the purification procedure because they are removed by extraction with phenol.
DIETHYLPYROCARBONATE
DEPC is used in molecular cloning to inactivate trace amounts of RNases that may contaminate solutions, glassware, and plasticware that are to be used for the preparation of nuclear RNA or mRNA (Penman et al. 1971; Williamson et al. 1971). DEPC is a highly reactive alkylating agent that destroys the enzymatic activity of RNase chiefly by ethoxyformylation of histidyl groups (see Fig. 1).
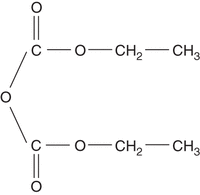
Structure of diethylpyrocarbonate.
Glassware and plasticware should be filled with a solution of 0.1% DEPC in H2O and allowed to stand for 1 h at 37°C or overnight at room temperature. Rinse the items several times with DEPC-treated H2O, then autoclave them for 15 min at 15 psi (1.05 kg/cm2) on liquid cycle.
In aqueous solution, DEPC hydrolyzes rapidly to CO2 and ethanol, with a half-life in phosphate buffer of ∼20 min at pH 6.0 and 10 min at pH 7.0. This hydrolysis is greatly accelerated by Tris and other amines, which themselves become consumed in the process. DEPC therefore cannot be used to treat solutions that contain these buffers. Samples of DEPC that are free of nucleophiles (e.g., H2O and ethanol) are perfectly stable, but even small amounts of these solvents can cause complete conversion of DEPC to diethylcarbonate. For this reason, DEPC should be protected against moisture. Store it in small aliquots in dry conditions, and always allow the bottle to reach ambient temperature before opening it.
Although H2O purified through well-maintained, modern reverse-osmosis systems is free of RNase (Huang et al. 1995), poorly maintained purification systems may become contaminated by microbial growth. This situation commonly occurs in large centralized systems with many meters of piping and storage vats in which H2O can stagnate. In such cases, it may be necessary to generate DEPC-treated H2O by treatment with 0.1% DEPC for 1 h at 37°C and autoclaving for 15 min at 15 psi (1.05 kg/cm2) on liquid cycle.
Other Uses of DEPC
In addition to reacting with histidine residues in proteins, DEPC can form alkali-labile adducts with the imidazole ring N7 of unpaired purines, resulting in cleavage of the glycosidic bond and generation of an alkali-labile abasic site (for review, see Ehrenberg et al. 1976). Because of its high reactivity and specificity, DEPC has been used as a chemical probe of secondary structure in DNA and RNA (e.g., see Peattie and Gilbert 1980; Herr 1985). Unpaired adenine residues are strongly reactive (Leonard et al. 1970, 1971), as are guanine residues in Z-DNA (Herr 1985; Johnston and Rich 1985). A diminution in the reactivity of purines with DEPC can therefore be used to measure binding between Z-DNA and specific proteins (Runkel and Nordheim 1986).
Problems in Using DEPC
Removal of DEPC by thermal degradation generates small amounts of ethanol and CO2, which can increase the ionic strength and lower the pH of unbuffered solutions. DEPC can carboxymethylate unpaired adenine residues in RNA. mRNAs that have been exposed to DEPC are translated with reduced efficiency in in vitro protein-synthesizing systems (Ehrenberg et al. 1976). However, the ability of DEPC-treated RNA to form DNA–RNA or RNA–RNA hybrids is not seriously affected unless a large fraction of the purine residues have been modified.
Footnotes
-
From the Molecular Cloning collection, edited by Michael R. Green and Joseph Sambrook.








