
Recombineering 101: Making an in-Frame Deletion Mutant
- 1Université Paris-Saclay, CEA, CNRS, Institut de Biologie Intégrative de la Cellule (I2BC), 91190 Gif-sur-Yvette, France
- 2Departamento de Genética, Facultad de Biología, Universidad de Sevilla, 41080 Sevilla, Spain
- ↵3Correspondence: lionello.bossi{at}i2bc.paris-saclay.fr
Abstract
DNA recombineering uses phage λ Red recombination functions to promote integration of DNA fragments generated by polymerase chain reaction (PCR) into the bacterial chromosome. The PCR primers are designed to have the last 18–22 nt anneal on either side of the donor DNA and to carry 40- to 50-nt 5′ extensions homologous to the sequences flanking the chosen insertion site. The simplest application of the method results in knockout mutants of nonessential genes. Deletions can be constructed by replacing a portion or the entirety of a target gene with an antibiotic-resistance cassette. In some commonly used template plasmids, the antibiotic-resistance gene can be coamplified with a pair of flanking FRT (Flp recombinase recognition target) sites that, following insertion of the fragment into the chromosome, allow excision of the antibiotic-resistance cassette via the activity of the site-specific Flp recombinase. The excision step leaves behind a “scar” sequence comprising an FRT site and flanking primer annealing sequences. Removal of the cassette minimizes undesired perturbations on the expression of neighboring genes. Even so, polarity effects can result from the occurrence of stop codons within, or downstream of, the scar sequence. These problems can be avoided by the appropriate choice of the template and by designing primers so that the reading frame of the target gene is maintained past the deletion end point. This protocol is optimized for use with Salmonella enterica and Escherichia coli.
MATERIALS
Reagents
Agarose (Fisher BP160-500)
Ampicillin stock solution (1000×)
Arabinose (20%), sterile
Bacterial strain to be used for mutagenesis (Salmonella enterica or Escherichia coli)
DMSO (100%; Thermo Scientific F-515; supplied with Phusion High-Fidelity DNA polymerase)
dNTP mix (10 mm each; Invitrogen 18427013)
Ethanol (95%)
Glycerol (10%), ice-cold, sterile
H2O (PCR grade)
LB 1.5% Agar Plates without and with Antibiotic Supplements
-
Prepare LB plates with no antibiotic as described in the recipe.
-
Prepare LB-Amp plates with 100 µg/mL ampicillin using Ampicillin Stock Solution (1000×). Store LB-Amp plates for up to 3 mo at 4°C.
-
Prepare LB-Kan plates with 50 µg/mL kanamycin using Kanamycin Stock Solution. Store LB-Kan plates for up to 6 mo at 4°C.
Lysogeny broth (LB; Lennox Formulation)
-
Where not specified, the LB used throughout this protocol is LB-Lennox.
Orange 6× DNA loading dye (Thermo Scientific R0631)
Phusion 5× GC buffer (Thermo Scientific F-519; supplied with Phusion High-Fidelity DNA polymerase)
Phusion High-Fidelity DNA polymerase (2 U/µL, Thermo Scientific F-530)
Plasmid pCP20 (FLP recombinase delivery plasmid; ts for replication; Cherepanov and Wackernagel 1995)
Plasmid pKD13 (PCR template plasmid; Datsenko and Wanner 2000)
Plasmid pKD46 (λ Red delivery plasmid; ts for replication; Datsenko and Wanner 2000)
Primer-1: (N)40–50GATCCGTCGACCTGCAGTTC (forward recombineering primer)
-
(N)40–50 denotes the 40- to 50-nt sequence immediately 5′ to the insertion/deletion boundary in the target gene.
Primer-2: (N)40–50GTGTAGGCTGGAGCTGCTT (reverse recombineering primer)
-
(N)40–50 denotes the 40- to 50-nt sequence complementary to the sequence immediately 3′ to the insertion/deletion boundary in the target gene.
Primer-3: (N)18–22 (verification primer)
-
(N)18–22 denotes an 18- to 22-nt sequence anywhere between 100 and 200 nt upstream of the insertion/deletion boundary in the target gene.
Primer-4: (N)18–22 (verification primer)
-
(N)18–22 denotes an 18- to 22-nt sequence complementary to a sequence anywhere between 100 and 200 nt downstream from the insertion/deletion boundary in the target gene.
Primer-KA (kan anti): CGGTTCGCTTGCTGTCCATA (verification primer)
-
This primer anneals upstream of the kan gene (antisense orientation).
Primer-KS (kan sense): GCCTGCTTGCCGAATATCAT (verification primer)
-
This primer anneals within the kan gene (sense orientation).
QIAquick PCR Purification Kit (QIAGEN 28106)
Quick load 100-bp (New England Biolabs N0467) and 1-kb ladders (New England Biolabs N0468)
SYBR Safe DNA Gel Stain (Invitrogen S33102)
TAE buffer (10×; Invitrogen 15558-026)
Taq DNA Polymerase 5000 U/mL, with ThermoPol buffer (New England Biolabs M0267)
Equipment
Centrifuge (refrigerated) with rotor that accommodates 50-mL tubes (e.g., Beckman Coulter Avanti, J series, with JA25.50 rotor)
Centrifuge bottles, polycarbonate with caps, 50-mL (Beckman Coulter 357002)
Culture tubes, glass (∼15-mm × 150-mm, diameter × length) with plastic caps, sterile
Electrophoresis gel casting and running apparatus
Electrophoresis power supply
Erlenmeyer flask (250-mL), sterile
Gel apparatus (tray, comb, and gel box)
Gel imager
Glass spreader (made from a glass rod) or glass plating beads
Incubator(s) (at 30°C and 37°C)
Microcentrifuge tubes, 1.5-mL (Eppendorf 022363743) and 2.0-mL (Eppendorf 022363344), autoclaved
Micropipettes (P2, P10, P20, P200, P1000)
MicroPulser electroporation cuvettes, 2-mm gap (BioRad 1652082)
MicroPulser electroporator (BioRad 1652100)
Microwave oven
NanoDrop spectrophotometer
Pasteur pipettes
PCR microcentrifuge tubes (0.2-mL)
Pipettor or pipetting device for serological pipettes
Plain wood applicator sticks, inoculators, sterile (Fisher Scientific 23-400-102)
Racks of micropipette tips, sterilized (10- and 200-µL, and 1-mL)
Serological pipettes (1-, 2-, 5-, and 10-mL), sterile
Shaking incubator(s) (orbital, at 30°C and 37°C)
Thermocycler
Wooden round toothpicks with sharp points (Fisher Scientific 504180), sterile
METHOD
-
Figure 1 shows an overview of the recombineering procedure.
-
This protocol is derived from the original procedure of Datsenko and Wanner (2000) with a few modifications. This protocol uses template plasmid pKD13 carrying an FRT-kan-FRT amplification cassette. In this plasmid, the use of primers annealing immediately adjacent to the FRT sites leaves a scar of 74 bp that is free of stop codons in two out of the three reading frames (Fig. 1A). In-frame deletions can be generated by placing the upstream and downstream insert junctions in the −1 and +1 frames, respectively (see Fig. 1B).
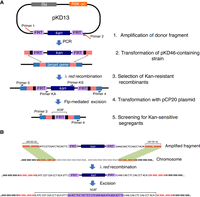
Schematic overview of the recombineering procedure. (A) Relevant steps involved. Regions of sequence homology between the donor fragment and chromosomal DNA are shown in salmon red. (B) Insertion frame requirements for reading frame maintenance following kan cassette excision.
Prologue
-
1. Design the 5′ extensions of recombineering primers (Primer-1 and Primer-2) according to the size and position of the desired deletion, respecting the requirement for reading frame maintenance. Design external verification primers (Primer-3 and Primer-4) using any of the available online tools (e.g., “Primer3”; Rozen and Skaletsky 2000). Order the oligonucleotides.
-
2. Introduce plasmid pKD46 in the strain to be used for the mutagenesis according to the quick transformation protocol (see Protocol: Quick Transformation with Plasmid DNA [Figueroa-Bossi et al. 2022]). Plate electroporated bacteria using a glass spreader or glass plating beads on an LB-Amp plate and incubate for 16–24 h at 30°C in an incubator.
-
3. On the next day, use toothpicks to streak a transformant colony on an LB-Amp plate. Incubate for 16–24 h at 30°C in an incubator.
-
Single colonies from this “purification streak” will constitute the source of recombineering-ready cells. The plate can be kept for a period of 3–4 wk at 4°C–6°C. For long-term use of the strain, consider making a 15% glycerol stock of the first culture grown up. This stock can be stored at −80°C, where cells will be viable and stable for years.
-
Day 1
Making the Donor Fragment
-
4. Assemble the components of the amplification reaction (50 µL in a 200-µL PCR microtube) as specified below. After adding the enzyme, mix gently by pipetting the liquid up and down a few times with a P200 micropipette.

-
Of the two buffers provided with the Phusion enzyme, Buffers HF and GC, we found the latter to give more consistent results and higher amplification yields under the conditions of this protocol, especially if used in combination with DMSO.
-
-
5. Set the PCR program in the thermocycler as follows:

-
6. Place the tube in the thermocycler and start the reaction.
-
7. While the PCR is running, prepare an agarose gel as follows.
-
i. Melt agarose in 1× TAE in the microwave oven to make a 1% gel.
-
ii. Add SYBR Safe DNA stain (to a 1/10,000 dilution).
-
iii. Cast the gel and allow it to solidify.
-
-
8. Once the amplification is finished, place the PCR tubes in an ice bucket. Withdraw 2 µL aliquots, mix them with 3 µL H2O and 1 µL 6× Orange loading dye each, and load them on the gel along with 0.5 µg of a 1-kb ladder.
-
9. Migrate for 15–30 min at 6 V/cm in 1× TAE buffer. Examine the gene under UV light (312-nm) and confirm the presence of a ∼1.4-kb band.
Purifying the PCR Fragment
-
This step uses the QIAquick PCR clean-up kit. Wear gloves as buffers in the kit contain harmful guanidine hydrochloride and isopropanol. Discard used buffers properly. Perform Steps 10–20 at room temperature.
-
10. Transfer the contents of the tubes from Step 8 to a 1.5-mL microcentrifuge tube.
-
11. Add 250 µL (5× volume) of buffer PB. Mix well by pipetting up and down a few times with a P1000 micropipette.
-
12. Load a QIAquick spin column.
-
13. Centrifuge at 13,000g in a microcentrifuge for 30 sec.
-
14. Discard the flowthrough but keep the column in the same collection tube.
-
15. Add 750 µL of buffer PE and centrifuge at 13,000g for 30 sec.
-
16. Discard the flowthrough but keep the column in the same collection tube.
-
17. Centrifuge again for 1 min to completely dry the column.
-
18. Place the column on a clean 1.5-mL microcentrifuge tube.
-
19. Add 30 µL of H2O to the center of the QIAquick column resin and let the liquid soak for 5 min.
-
20. Centrifuge at 13,000g for 1 min and collect the flowthrough.
-
21. Measure concentration in the Nanodrop and store the DNA at −20°C.
Preparation of Electro-Competent Cells
-
22. Pick a single colony from Step 2 with an applicator stick and inoculate a 2- to 3-mL culture in LB supplemented with 100 µg/mL of ampicillin in a culture tube. Grow overnight at 30°C with 170 rpm agitation.
Day 2
-
23. Transfer 0.35 mL of the overnight culture into a 250-mL Erlenmeyer flask containing 35 mL of LB supplemented with 100 µg/mL ampicillin and 0.1% arabinose. Place the flask in an orbital shaker incubator and resume growth at 30°C with 170 rpm agitation.
-
24. Precool the centrifuge for 50-mL tubes and the microcentrifuge with their respective rotors at 4°C.
-
25. Harvest cells after 3 h of growth at 30°C by centrifuging at 6800g for 4 min at 4°C in a sterile polycarbonate 50-mL centrifuge bottle.
-
At this time, the optical density of the culture at 600 nm (OD600) is typically between 0.6 and 0.7 (∼2 × 108 cfu/mL).
-
-
26. Pour off the supernatant and resuspend the pellet in 35 mL of ice-cold, sterile 10% glycerol as follows: add 5 mL first and detach the pellet by pipetting gently up and down on the pellet side of the tube. When cell suspension is homogeneous (no visible clumps), bring volume up to 35 mL by adding 30 mL of ice-cold 10% glycerol. Mix by inverting the centrifuge tube three or four times.
-
27. Repeat the centrifugation at 6800g for 4 min at 4°C.
-
28. Pour off the supernatant and resuspend the pellet in 1.5 mL of ice-cold 10% glycerol pipetting the liquid up and down as before. Transfer the cell suspension to a 2-mL microcentrifuge tube.
-
29. Pellet the cells again, this time in the precooled microcentrifuge for 1.5 min at 9000g at 4°C.
-
30. Pour off the supernatant immediately after the centrifuge stops. Tap the tube opening on tissue paper several times to discard as much liquid as possible.
-
31. Gently resuspend the pellet in 850 µL of ice-cold 10% glycerol. Bring the volume to 1.7 mL by adding 850 µL more of ice-cold 10% glycerol. Mix by inverting the tube two or three times.
-
32. Repeat the centrifugation for 1.5 min at 4°C at 9000g.
-
33. Repeat Step 30.
-
34. Repeat Step 31.
-
35. Repeat Step 29.
-
36. Repeat Step 30.
-
37. Perform a last short (15-sec) centrifugation and remove the residual liquid with a 200-µL micropipette, immediately after the centrifuge stops.
-
38. Resuspend the pellet in 80 µL of ice-cold 10% glycerol. Use 45–50 µL for electroporation.
-
Taking into account the volume of the cell pellet, the final volume of the cell suspension is usually >100 µL, which is enough material to carry out two electroporations (if more than one mutant is being constructed). However, we do not recommend deep-freezing the leftover cells for use at another time, as recombineering efficiency drops dramatically after storage.
-
Electroporation and Selection of Recombinants
-
39. In a cold microcentrifuge tube, mix 45–50 µL of the electrocompetent cell suspension with 5 µL of the PCR-amplified, QIAGEN-purified DNA fragment from Step 21 (typically ∼0.5–1 µg total). Place the mix in a chilled electroporation cuvette (1- or 2-mm gap), gently tap the cuvette on a solid surface to make sure that the liquid is at the bottom and that no bubbles are present.
-
40. Follow the instructions from the electroporator manufacturer to setup electroporation parameters corresponding to the cuvette gap.
-
Typically, the parameters are 2.5 kV, 200Ω, and 25 µF for a 0.2 cm electroporation cuvette.
-
-
41. Insert the cold cuvette in the electroporator and apply pulse.
-
42. Immediately after the pulse, put 1 mL of LB into the cuvette with a Pasteur pipette and dilute cells by gently pipetting the liquid up and down. Transfer the suspension to a culture tube.
-
43. Incubate the cells for at least 1.5 h at 37°C with 170 rpm shaking.
-
This outgrowth period is needed for bacteria to recover from the high voltage pulse and to express the Kan-resistance gene.
-
-
44. Spread 0.1 mL of the culture on an LB-Kan plate using a glass spreader or glass beads and incubate overnight at 37°C.
Day 3
-
45. Using toothpicks, streak 2–4 colonies on an LB-Kan plate. Incubate overnight at 37°C.
-
Growth at 37°C causes the loss of the pKD46 plasmid.
-
See Troubleshooting.
-
Day 4
Strain Verification by Colony PCR
-
Check at least two independent isolates.
-
46. Prepare 0.2-mL PCR tubes with 50 µL of H2O.
-
47. Pick a small lump of each colony with a sterile micropipette tip (without the pipette attached) and resuspend the material in the H2O.
-
48. Attach a micropipette to the tip and pipette the liquid up and down until no clumps are visible. The suspension should look slightly turbid.
-
49. Put the tubes in a thermocycler and incubate for 10 min at 98°C.
-
50. Centrifuge tubes (with PCR-tube adaptors) for 2 min at room temperature in a microcentrifuge at maximum speed. A small white pellet should be visible.
-
51. Collect the supernatant with a P200 micropipette. Use a 5-µL aliquot for colony PCR. If desired, freeze the remainder of the supernatant.
-
52. Prepare the amplification reaction mixture as specified below.
-
The recipe below is for two 40-µL reactions (one for each of two clones being tested). For each clone, the protocol calls for separate verifications of the left and right insert junctions. In the table below, forward and reverse primers are Primer-3 and Primer-KA for the left junction, and Primer-KS and Primer-4 for the right junction. If the length of the deleted material differs considerably from the insert size, one may decide to simply check for the presence of the insert by using Primers-3 and Primer-4 as forward and reverse primers (see Fig. 1A).
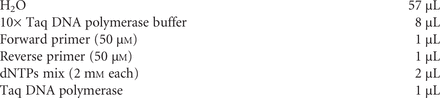
-
To obtain 2 mm dNTPs, make a 1:5 dilution of the 10 mm dNTP solution used for PCR with the Phusion enzyme.
-
Divide the mix into two tubes (35 µL in each) and add 5 µL of each colony lysate. Mix gently.
-
-
53. Set the PCR program in the thermocycler as follows:

-
54. Place tubes in the thermocycler and start the reaction.
-
55. While the PCR is running, pour a 1% agarose gel with SYBR Safe added as in Step 7.
-
56. Once amplification is finished, place the microtubes in an ice bucket. Withdraw 2-µL aliquots, mix with loading buffer (as described in Step 8) and load on the gel along with 0.5 µg of a 100-bp DNA ladder.
-
57. Migrate for 15–30 min at 6 V/cm. Examine the gel under UV light (312-nm). If bands of the predicted sizes are present, proceed to the excision of the kan cassette.
-
If desired, start using the mutant obtained at this stage directly for further studies. If doing so, verify that the sequences at the boundaries of the cassette are as expected. To do so, purify the fragments obtained in Step 56 (as described in Steps 10–20) and send them to a Sanger sequencing service along with Primer-3 (left junction fragment) and Primer-4 (right junction fragment).
-
Excision of the kan Cassette
-
58. Inoculate a 2-mL culture with a single colony from an isolate that checked out in Step 57. Grow in LB at 37°C with 170 rpm shaking.
-
59. Precool a microcentrifuge with rotor at 4°C.
-
60. After 2–3 h of growth (OD600 anywhere between 0.5 and 0.8; with practice one can gauge the correct turbidity by eye), centrifuge the culture at 7500g for 1.5 min at 4°C and prepare electrocompetent cells as outlined in Protocol: Quick Transformation with Plasmid DNA (Figueroa-Bossi et al. 2022).
-
61. Electroporate cells with plasmid pCP20 DNA as described in Protocol: Quick Transformation with Plasmid DNA (Figueroa-Bossi et al. 2022).
-
62. Allow cells to recover by incubating for 1.5 h at 30°C with 170 rpm shaking.
-
63. Spread 100 µL of the transformed culture on an LB-Amp plate using a glass spreader or glass beads.
-
64. Incubate the plate for 16-24 h at 30°C.
Day 5
-
65. Using toothpicks pick two to four amp-resistant colonies and streak them on an LB-Amp plate.
-
This single-colony purification step allows segregation of Kan-sensitive clones resulting from Flp-recombinase-mediated excision of the kan cassette.
-
-
66. Incubate the plate for 16–24 h at 30°C.
Day 6
-
67. Choose 4 isolates and using toothpicks sequentially touch an LB-Kan plate and an LB-Amp plate (each divided in four sectors). Streak for single colonies.
-
68. Incubate the plates overnight at 37°C.
Day 7
-
69. Examine the plates. Most, in not all, of the four streaks should not have grown on the Kan-supplemented plate, indicative of the loss of the Kan-resistance cassette.
-
At the same time growth on LB at 37°C will have selected for the loss of pCP20 (ts for replication).
-
-
70. Choose one of the clones that checks out phenotypically and perform colony PCR verification as described in Steps 46–57 using Primer-3 and Primer-4.
-
71. Purify the PCR fragment as in Steps 10–20 and send it to a Sanger sequencing service along with either Primer-3 or Primer-4.
TROUBLESHOOTING
Problem (Step 45): On day 3 there are no colonies on the plate.
Solution: If no colonies are visible on day 3 (and there is no reason to expect that the mutant being constructed has a slow growth rate), chances are that something went wrong with the experiment. Do not give in to the temptation of analyzing colonies that come up at later times, which is likely a waste of time. If desired, try adding 1–2 mL of LB to the electroporated cell suspension leftover from the day before. Incubate in the shaker for 4–5 h and plate again. However, it is generally wiser to just back up and repeat the whole procedure again with fresh cells.
ACKNOWLEDGMENTS
Work in our laboratory was supported by the Centre National de la Recherche Scientifique (CNRS) and by the Agence Nationale de la Recherche (ANR-15-CE11-0024-03), France.
Footnotes
-
From the Experiments in Bacterial Genetics collection, edited by Lionello Bossi, Andrew Camilli, and Angelika Gründling.








