
Using CRISPR–Cas9-Based Methods for Genome Editing in Staphylococcus aureus
- 1Section of Molecular Microbiology and Medical Research Council Centre for Molecular Bacteriology and Infection, Imperial College London, London SW7 2AZ, United Kingdom
- 2School of Physical Science and Technology, ShanghaiTech University, Shanghai 201210 China
- 3Department of Laboratory Medicine and Pathology, School of Medicine, University of Washington, Seattle, Washington 98195, USA
- ↵4Correspondence: a.grundling{at}imperial.ac.uk; quanjiangji{at}shanghaitech.edu.cn; stevesal{at}uw.edu
Abstract
Chromosomal mutations and targeted gene deletions and inactivations in Staphylococcus aureus are typically generated using the allelic exchange method. In recent years, however, more rapid methods have been developed, often using CRISPR–Cas9-based systems. Here, we describe recently developed CRISPR–Cas9-based plasmid systems for use in S. aureus, and discuss their use for targeted gene mutation and inactivation. First, we describe how a CRISPR–Cas9 counterselection strategy can be combined with a recombineering strategy to generate gene deletions in S. aureus. We then introduce dead Cas9 (dCas9) and Cas9 nickase (nCas9) enzymes, and discuss how the nCas9 enzyme fused to different nucleoside deaminases can be used to introduce specific base changes in target genes. We then discuss how the nCas9-deaminase fusion enzymes can be used for targeted gene inactivation via the introduction of premature stop codons or by mutating the start codon. Together, these tools highlight the power and potential of CRISPR–Cas9-based methods for genome editing in S. aureus.
CRISPR–Cas9 PLASMID SYSTEMS FOR USE IN STAPHYLOCOCCUS AUREUS
In the last few years, several CRISPR–Cas-based plasmid systems have been developed for use in Staphylococcus aureus, which can be used to generate gene deletions or inactivations (Chen et al. 2017; Zhao et al. 2017; Gu et al. 2018; Penewit et al. 2018). These plasmids are usually based on the Streptococcus pyogenes CRISPR–Cas9 system. Using the CRISPR–Cas9 system, a site-specific double-strand break can be introduced in a gene of interest, which is lethal for S. aureus unless repaired. For the repair process, researchers typically use a double-stranded DNA-repair template with 1-kb homology regions upstream and downstream of the double-strand break. In this manner, gene deletions, point mutations, or even gene fusions can be incorporated into the repair template and then introduced into the chromosome.
Three elements are needed for successful double-strand cleavage. First, a 20-nt CRISPR RNA (crRNA), also referred to as a spacer RNA, is needed, which binds to a complementary chromosomal sequence and provides specificity to the system, dictating where the double-strand cleavage will take place. Second, a trans-acting RNA (tracrRNA), which interacts with the crRNA and binds to and recruits the Cas9 protein, is needed. And third, the Cas9 protein, an RNA-guided endonuclease that introduces the double-strand break, is required. To simplify the system, the crRNA and tracrRNA are often fused together to generate a single synthetic guide RNA (sgRNA), which contains, at its 5′ end, the variable 20-base crRNA (spacer RNA) and, at the 3′ end, the constant tracrRNA (Fig. 1). Another sequence feature required for this system to function and that needs to be present immediately adjacent to the spacer RNA-binding site, is a protospacer-adjacent motif (PAM) site. The PAM site is recognized by the Cas9 enzyme and contributes to the DNA target specificity (Fig. 1). In the case of the S. pyogenes Cas9 protein, the PAM-recognition site has the sequence “NGG.” This, therefore, creates a limitation, as double-strand breaks cannot be introduced at every position in the genome, as the variable space sequence needs to be designed to base pair with a DNA sequence that is adjacent to a PAM site (Fig. 1).
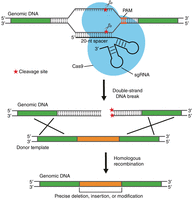
CRISPR–Cas9 genome-editing method. Schematic representation of how the CRISPR–Cas9 system can be used to introduce a site-specific double-strand break and how the break can be repaired. The Cas9 enzyme, bound to an appropriately designed synthetic guide RNA (sgRNA), will bind to the chromosome around a protospacer-adjacent motif (PAM) site and introduce a double-strand break. By providing a suitable double-strand DNA-repair template with long homology regions on both sides of the double-strand break site, the break will be repaired. In this manner, using a custom template, gene deletions, insertions, and single-base changes can be made. (Reprinted, with permission, from Chen et al. 2017, © American Chemical Society.)
For CRISPR–Cas9-mediated genetic manipulation of S. aureus, Ji and collaborators developed plasmid pCasSA (Fig. 2), which allows for the expression of (1) the gene-specific sgRNA from the S. aureus capA1 promoter, and (2) the S. pyogenes Cas9 protein from the strong rpsL promoter (Chen et al. 2017). The variable spacer and, hence, gene-targeting sequence, can be introduced between two BsaI restriction sites in pCasSA. The DNA-repair template is introduced into the same plasmid, between the XbaI and XhoI restriction sites, either using restriction enzymes and standard cloning procedures, or using the Gibson assembly method. To create a gene deletion, the spacer RNA is designed to target a specific gene, and a repair DNA template, comprising 1-kb DNA fragments upstream and downstream of the gene to be deleted, is inserted between the XbaI and XhoI sites on the same plasmid. The repair DNA fragment can be generated and introduced into the plasmid in a similar manner, as described, for instance, for the allelic exchange procedure in Protocol: Construction of a Staphylococcus aureus Gene Deletion Allelic Exchange Plasmid by Gibson Assembly and Recovery in Escherichia coli (Zeden et al. 2023a). Once the proper spacer sequences and the homologous repair template have been introduced into pCasSA, the plasmid is then transformed into S. aureus, and gene-deletion mutants are generated in a single step and usually obtained within 2 days. As such, generating gene deletions using the CRISPR–Cas9 system is significantly faster than the method traditionally used in S. aureus for generating gene deletions, the allelic exchange method (see Introduction: Allelic Exchange: Construction of an Unmarked In-Frame Deletion in Staphylococcus aureus [Zeden et al. 2023b]). The efficiency of obtaining transformants with the desired gene deletion, however, varies greatly among genes and, hence, additional methods for generating gene deletions or inactivations in S. aureus have been developed based on some aspects of the CRISPR–Cas9 system. For instance, a recombineering strategy combined with a CRISPR–Cas9 counterselection method has been established in S. aureus for the generation of gene deletions, and the principle of the method is described below. Information on CRISPR–Cas9 and how it can be used in combination with a cytidine or adenosine deaminase to generate point mutations in S. aureus is also discussed in this article, and the application of a CRISPR–Cas9-mediated method for introducing point mutations in a Vibrio cholerae phage is described in Protocol: CRISPR Gene Editing of a Virulent Bacteriophage ICP1 (Camilli 2023).
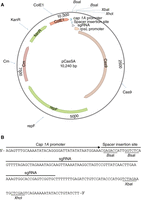
pCasSA plasmid. (A) Plasmid map of the Staphylococcus aureus CRISPR–Cas9 genome-editing plasmid pCasSA, with relevant features indicated. (B) DNA sequence showing the synthetic guide RNA (sgRNA) sequence with a spacer insertion site flanked by BsaI restriction sites and XbaI and XhoI sites for inserting the DNA repair template. (Reprinted, with permission, from Chen et al. 2017, © American Chemical Society.)
THE RECOMBINEERING CRISPR–Cas9 COUNTERSELECTION METHOD
There are numerous tools for introducing genetic modifications of interest in bacteria. Recombination-mediated genetic engineering (commonly referred to as “recombineering”) is a method that allows in vivo genetic engineering of the bacterial chromosome without the need for standard cloning techniques, and uses either a single-stranded DNA oligonucleotide or double-stranded DNA molecules generated by polymerase chain reaction (PCR). In this method, the in vitro–generated DNA molecule designed to generate the desired genomic alterations is introduced into a bacterium usually expressing a phage recombination system. The general principles behind recombineering of bacteria, and how they can be used to generate gene deletions, gene fusions, or point mutations in enteric bacteria, are described in detail in Introduction: DNA Recombineering Applications (Figueroa-Bossi et al. 2023) and references therein. Briefly, in enteric bacteria, the bacteriophage λ Red proteins are expressed in the target bacterium to promote efficient recombination between a PCR product or single-stranded oligonucleotide and the bacterial chromosome, to produce the desired genomic alteration. Recently, a recombineering system has also been developed for use in the Gram-positive bacterium S. aureus (Penewit et al. 2018). Here, the recombination between a single-stranded oligonucleotide and the chromosomal DNA is catalyzed by expressing the Enterococcus faecalis recombinase enzyme EF2132 (Penewit et al. 2018). Recombination efficiency in this system, however, is relatively low compared to those achievable in enteric organisms using the λ Red proteins. Therefore, recombineering techniques in S. aureus are combined with a counterselection strategy, wherein bacteria that have not undergone the recombination process are selected against by introducing a lethal CRISPR–Cas9-mediated double-strand DNA break. This method of using recombineering with a single-stranded oligonucleotide and subsequent CRISPR–Cas9 counterselection for generating gene deletions in S. aureus was first described in 2018 (Penewit et al. 2018). In this collection, we provide an example of such an approach, which we describe in Protocol: Oligonucleotide Design and Construction of a Gene-Targeting CRISPR–Cas9 Plasmid in Escherichia coli for Generating a Gene-Deletion Strain in Staphylococcus aureus (Gründling and Salipante 2023a) and Protocol: Introduction of a Recombineering Oligonucleotide and a CRISPR–Cas9 Gene-Targeting Plasmid into Staphylococcus aureus for Generating a Gene-Deletion Strain (Gründling and Salipante 2023b). In these examples, and similar to what is described above for plasmid pCasSA, a specific plasmid, termed pCas9-counter, was developed to allow for the introduction of a site-specific double-strand break using CRISPR–Cas9 and features an appropriate spacer sequence. In the example described in the aforementioned protocols, a double-strand break is introduced in the S. aureus geh gene, allowing for counterselection of S. aureus strains with an intact geh gene (i.e., strains with a wild-type [WT] copy of the geh gene will be subjected to a double-strand break without repair, and will thus be selected against). In this case, and instead of cloning a large DNA fragment with the 1-kb homology region into the plasmid to serve as a repair template, a single-stranded 90-mer DNA oligonucleotide is introduced by electroporation alongside the CRISPR–Cas9 plasmid into an S. aureus strain expressing the E. faecalis recombinase EF2132. The oligonucleotide is designed in such a manner that ∼45 bases match the sequences upstream, and ∼45 bases match the sequences downstream the genomic region that is subject to the CRISPR–Cas9-mediated double-strand break. Using such an oligonucleotide, a gene deletion can be introduced around the CRISPR–Cas9 cleavage site (Fig. 3). In this system, the single-stranded DNA oligonucleotide serves as template for a recombineering event promoted by the recombinase EF2132. As mentioned, the CRISPR–Cas9 system provides a counterselection mechanism to kill bacteria that have not undergone recombineering, that is, WT bacteria with an intact copy of the gene intended to be deleted (in our example, the geh gene). A schematic representation of this process is shown in Figure 3.
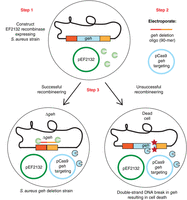
Schematic representation of the recombineering CRISPR–Cas9 counterselection method described in this collection to create gene deletions in Staphylococcus aureus. Step 1: An S. aureus strain containing plasmid pCN-EF2132tet (in the schematic, indicated as pEF2132) for expression of the Enterococcus faecalis recombinase EF2132 is constructed. Step 2: A 90-mer single-stranded DNA oligonucleotide containing sequences flanking the target gene (in this example, geh) is electroporated into the S. aureus strain from Step 1 alongside a CRISPR–Cas9 plasmid also targeting the target gene. Step 3: Upon successful recombineering using the oligonucleotide, a target-gene deletion strain is obtained (Δgeh in our example, left cell). If recombineering with the oligonucleotide does not take place, a Cas9-mediated double-strand break is introduced into the gene to be deleted, killing such bacteria with a wild-type allele of the gene (right cell).
In this workflow, and as a first step, an S. aureus strain is generated that contains plasmid pCN-EF2132tet, allowing for constitutive expression of the E. faecalis recombinase EF2132. As a second step, an appropriately designed oligonucleotide (in this example, an oligonucleotide for generating a geh gene deletion) is electroporated along with the pCas9-counter plasmid (in this example, containing a spacer sequence targeting the geh gene) into the EF2132 recombinase-expressing S. aureus strain. A successful recombineering event will lead to the generation of a gene-deletion strain (Fig. 3, Step 3). On the other hand, bacteria that did not successfully undergo the recombineering event and still contain an intact copy of the gene of interest (the geh gene in this example), will be killed through the introduction of a CRISPR–Cas9-mediated double-strand break in the gene of interest (Fig. 3, Step 3). Because the two plasmids used for the recombineering and the CRISPR–Cas9 counterselection have temperature-sensitive origins of replication in S. aureus, the S. aureus strains are grown at low temperature (typically 30°C–32°C) for the recombineering process. However, once the desired mutation has been obtained, the strain can easily be cured of the plasmids by propagating the bacteria for a few generations at a higher temperature (typically 37°C–42°C), which is not permissive for plasmid replication. The loss of the plasmids can be confirmed by patching bacteria on agar plates containing the appropriate antibiotics.
In contrast to the λ Red system, no obvious difference in recombineering efficiency is observed for oligonucleotides complementary to either the leading or lagging DNA replication strand and, hence, it is advisable to design and test oligonucleotides for both strands. An oligonucleotide with a length of 90 bases was found to be optimal for the recombineering process, as it was observed that the efficiency decreased when the oligonucleotide length was increased to 100 bases or decreased to 70 bases (Penewit et al. 2018). Finally, incorporation of several phosphorothioate bonds at the 5′ end of the oligonucleotide has been shown to improve recombineering efficiency, presumably by preventing degradation of the oligonucleotide by exonucleases. Furthermore, and similarly to what has been reported for Escherichia coli, the system is more efficient for creating small gene deletions than large gene deletions (Penewit et al. 2018). In our specific example (see below), a 55-bp out-of-frame deletion in the geh gene, centered around a selected CRISPR–Cas9 cut site, will be generated at the beginning of the geh gene.
Although not performed as part of the examples provided in this collection (see below), oligonucleotides can also be designed to introduce base changes instead of gene deletions; however, some constraints apply. The recombineering oligonucleotide should be designed in such a manner that the PAM site is inactivated through the recombineering event, and the mutated sequence can no longer be recognized and cleaved by the Cas9 enzyme. Furthermore, in addition to the desired point mutation, additional silent mutations will need to be introduced as part of the oligonucleotide sequence, so that the point mutation is retained and can escape recognition and repair by the DNA mismatch-repair system, which recognizes and repairs erroneous DNA insertions, deletions, and misincorporation of bases during the DNA replication process. Further details on oligonucleotide design can be found elsewhere (Penewit et al. 2018; Penewit and Salipante 2020).
In this collection, we provide step-by-step instructions on how to construct an S. aureus gene-deletion strain using the recombineering and CRISPR–Cas9 counterselection method. We describe the entire approach in two protocols, which need to be performed consecutively (see Protocol: Oligonucleotide Design and Construction of a Gene-Targeting CRISPR–Cas9 Plasmid in Escherichia coli for Generating a Gene-Deletion Strain in Staphylococcus aureus [Gründling and Salipante 2023a]; and Protocol: Introduction of a Recombineering Oligonucleotide and a CRISPR–Cas9 Gene-Targeting Plasmid into Staphylococcus aureus for Generating a Gene-Deletion Strain [Gründling and Salipante 2023b]). In the first protocol, we describe the design of the recombineering oligonucleotide, as well as the construction of the CRISPR–Cas9 gene-targeting plasmid in E. coli (see Protocol: Oligonucleotide Design and Construction of a Gene-Targeting CRISPR–Cas9 Plasmid in Escherichia coli for Generating a Gene-Deletion Strain in Staphylococcus aureus [Gründling and Salipante 2023a]). In the second protocol, we describe the process for generating and verifying the actual gene deletion in S. aureus (see Protocol: Introduction of a Recombineering Oligonucleotide and a CRISPR–Cas9 Gene-Targeting Plasmid into Staphylococcus aureus for Generating a Gene-Deletion Strain [Gründling and Salipante 2023b]). In these protocols, we use the S. aureus geh gene as an example.
Colony PCR is used in multiple steps in the workflow. In addition to the colony PCR method for S. aureus described in Protocol: Introduction of a Recombineering Oligonucleotide and a CRISPR–Cas9 Gene-Targeting Plasmid into Staphylococcus aureus for Generating a Gene-Deletion Strain (Gründling and Salipante 2023b), the collection also features an article that includes another protocol for colony PCR for consideration (see Protocol: Allelic Exchange Procedure in Staphylococcus aureus [Zeden et al. 2023c]).
dCAS9 and nCAS9 ENZYMES
An enzymatically inactive or “dead” Cas9 (dCas9) enzyme is used in many systems to silence gene expression and to create roadblocks for the transcription machinery, and such systems have now also been developed for use in S. aureus (Chen et al. 2017; Zhao et al. 2017). By mutating the two key active site residues in Cas9, Asp10 and His840, to alanines, an enzymatically dead Cas9 enzyme, dCas9, is generated. This dCas9 enzyme can still be targeted and bind to a specific DNA site on the chromosome. However, instead of introducing a double-strand break, the protein will now serve as a roadblock for transcription. For this system to work, the spacer sequences need to be designed in such manner that the spacer RNA binds to the DNA strand that is not used as a template for transcription (non-template strand). Two S. aureus plasmids that encode the dCas9 enzyme are, for instance, plasmid pSD1, developed by Sun and coworkers (Zhao et al. 2017), and plasmid pCasiSA, developed by Ji and coworkers (Chen et al. 2017). In addition to “dead” Cas9 enzymes, researchers have also generated Cas9 variants, which, although they cannot introduce a double-strand break, can still nick one DNA strand. One such nicking enzyme is nCas9, which was generated by mutating only the Asp10 to alanine. Like the Cas9 enzyme, the nCas9 enzyme can be targeted to specific sites on the chromosome by expressing appropriately designed sgRNAs, but instead of generating a double-strand break, the nCas9 enzyme will only cleave one DNA strand.
USING nCas9-DEAMINASE FUSION ENZYMES FOR GENOME EDITING
For genome-editing purposes, systems have been developed in which the nCas9 enzyme is fused to either the deaminase enzyme APOBEC1, which allows for cytosine base editing, or an adenosine deaminase ABE7.10, which allows adenine base editing (Gaudelli et al. 2017; Gu et al. 2018; Zhang et al. 2020). In Figure 4C, we show the mechanism by which the cytidine deaminase APOBEC1 functions to convert cytosines (C) to uracils (U), and how the CRISPR–APOBEC1–nCas9 system works to introduce a nick as well as a C-to-T conversion at a specific chromosomal site. Similarly, in Figure 4D, we show how the adenosine deaminase ABE7.10 converts adenines (A) to hypoxanthines (I), and how the CRISPR–ABE7.10–nCas9 functions to introduce A-to-G conversions.
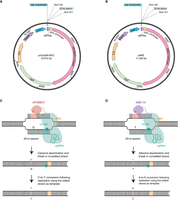
Staphylococcus aureus plasmids and schematic representation of genome-editing methods using nCas9-deaminase fusion enzymes. (A) Map of the S. aureus pnCasSA-BEC plasmid used for the expression of the APOBEC1–nCas9 fusion enzyme, with relevant features indicated. (B) Map of the S. aureus pABE plasmid used for the expression of the ABE7.10–nCas9 fusion enzyme, with relevant features indicated. (C) Schematic representation of the process leading to a C-to-T conversion using the APOBEC1–nCas9 fusion enzyme. The APOBEC1–nCas9 fusion enzyme bound to an appropriately designed synthetic guide RNA (sgRNA) will bind to the chromosome around a protospacer-adjacent motif (PAM) site. The nCas9 enzyme will introduce a nick in the bottom DNA strand, whereas the APOBEC1 deaminase will convert an appropriately spaced cytosine (C) in the top strand to a uracil (U). This will lead to a C-to-T base conversion during DNA replication. (D) Schematic representation of the process leading to an A-to-G conversion using the ABE7.10–nCas9 fusion enzyme. The ABE7.10–nCas9 enzyme bound to an appropriately designed sgRNA will bind to the chromosome around a PAM site. The nCas9 enzyme will introduce a nick in the bottom DNA strand and the ABE7.10 deaminase will then convert an appropriately spaced adenine (A) in the top strand to a hypoxanthine (I). This will result in an A-to-G conversion during DNA replication. Plasmid maps shown in panels A and B were generated using SnapGene.
For expression of the nCas9–APOBEC enzyme in S. aureus, plasmid pCasSA (Chen et al. 2017), discussed above, was modified to generate plasmid pnCasSA-BEC (Fig. 4A; Gu et al. 2018). In plasmid pnCasSA-BEC, the single-guide RNA is expressed from the S. aureus cap1A promoter and the APOBEC1–nCas9 fusion protein is expressed from the rpsL promoter (Fig. 4A). In this system, the nCas9 enzyme will introduce a nick into one of the DNA strands and the fused cytidine deaminase enzyme will convert nearby cytosines to uracils on the other strand. The cytosine will need to be present at position 4–8 in the spacer region (red bases in Fig. 4C), and, hence, 17–13 bases upstream of the “NGG” PAM site. Here, it is also important to note that conversion of a cytosine at position 4 is less efficient than conversion of a cytosine at positions 5–8. Depending on the open reading frame and spacing from the PAM site, CAA (Gln), CAG (Gln), or CGA (Arg) codons in the plus-strand of genes can be converted to TAA, TAG, or TGA stop codons, respectively. Similarly, a TGG (Trp) codon can be converted to a TAA stop codon when targeting the minus strand. Although it is possible to edit many cytosines within a bacterial genome, introducing actual stop codons is less frequent. Nevertheless, using this system, it is possible to introduce gene-inactivating mutations in many genes.
For the expression of the ABE7.10–nCas9 enzymes in S. aureus, plasmid pABE was developed (Fig. 4B; Zhang et al. 2020). In plasmid pABE, the single-guide RNA is again expressed from the S. aureus cap1A promoter and the ABE7.10–nCas9 fusion protein is expressed from the rpsL promoter (Fig. 4B). As described above, the nCas9 enzyme will introduce a nick into one of the DNA strands and, in this case, the fused adenosine deaminase enzyme will convert a nearby adenine (A) to a hypoxanthine (I) on the other strand, which will result in A-to-G conversions during replication (Fig. 4D). The editable adenine bases will again need to be present at positions 4–8 in the spacer region (red bases in Fig. 4D), and, hence, 17–13 bases upstream of the “NGG” PAM site. Using the adenine base editor system, internal stop codons can be generated or ATG start codons can be mutated to ACG codons, hence leading to gene silencing.
To identify C-to-T conversions or A-to-G conversions that lead to the introduction of stop codons, a careful analysis of each gene is needed. For the S. aureus strains, Newman and MRSA252, all possible editable stop sites resulting from C-to-T conversions in the genome have been reported and published as part of a bioinformatic analysis presented in the original paper describing the method (Gu et al. 2018). Hence, for these and closely related S. aureus strains, editable stop sites can be looked up. Furthermore, bioinformatic pipelines and gene-analysis tools have now been developed to identify cytosine or adenosine bases that, when mutated, will lead to the introduction of a stop codon in a target gene (Yu et al. 2020). For instance, CRISPR–CBEI, a base editor–mediated gene analysis tool, can be used to identify editable bases and spacer sequences within genes (Yu et al. 2020). This tool can also be used to identify potential off-target effects by determining how closely a chosen spacer sequence matches other chromosomal sequences.
In this collection, we provide step-by-step instructions on how to introduce a stop codon into a target gene using the CRISPR–nCas9 and cytidine deaminase system in S. aureus, for gene inactivation. We use the S. aureus geh gene as an example, and describe the entire approach in two protocols, which need to be performed consecutively. In this example, two cytosines (C160, part of a CAA codon, and C712, part of a CAG codon) in the geh gene will be converted to thymines, which will lead to the introduction of TAA (C160) and TAG (C712) stop codons, respectively. Upon successful conversion of these cytosines to a thymine, lipase-negative S. aureus strains RN4220-geh(160stop) and RN4220-geh(712stop) will be generated. Specific examples are presented to better show the oligonucleotide design. In the first protocol, we describe the identification of editable cytosine sites in the gene of interest using the CRISPR–CBEI toolkit and the design of the spacer oligonucleotide. We also provide the steps for the construction of the CRISPR–nCasSA gene-targeting plasmid in E. coli (see Protocol: Identification of Editable Sites, Spacer Oligonucleotide Design, and Generation of the Gene-Targeting CRISPR–nCas9 Plasmid for Gene Disruption in Staphylococcus aureus Using the CRISPR–nCas9 and Cytidine Deaminase System [Gründling and Ji 2023a]). In the second protocol, we describe the process for generating and verifying the actual gene-inactivation mutant in S. aureus (see Protocol: Introduction of a CRISPR–nCas9 Gene-Targeting Plasmid into S. aureus for Gene Disruption [Gründling and Ji 2023b]). The strategy described in the aforementioned protocols can easily be adapted to generate stop codons in other genes. Additional procedures on how to use CRISPR–Cas9 technologies to generate mutations in S. aureus can be found in Chen and Ji (2020).
ACKNOWLEDGMENTS
The research in the Gründling laboratory is supported by the Wellcome Trust grant 210671/Z/18/Z/WT.
Footnotes
-
From the Experiments in Bacterial Genetics collection, edited by Lionello Bossi, Andrew Camilli, and Angelika Gründling.








