
Considerations for Using Phage Display Technology in Therapeutic Antibody Drug Discovery
- 1Ailux Biologics, Somerville, Massachusetts 02143, USA
- 2GlobalBio, Inc., Cambridge, Massachusetts 02138, USA
- ↵3Correspondence: maryann.pohl{at}xtalpi.com
Abstract
Phage display is a versatile and effective platform for the identification and engineering of biologic-based therapeutics. Using standard molecular biology laboratory techniques, one can create a highly diverse and functional antibody phage-displayed library, and rapidly identify antibody fragments that bind to a target of interest with exquisite specificity and high affinity. Here, we discuss key aspects for the development of an antibody discovery strategy to harness the power of phage display technology to obtain molecules that can successfully be developed into therapeutics, including target validation, antibody design goals, and considerations for preparing and executing phage panning campaigns. Careful design and implementation of discovery campaigns—regardless of the target—provides the best chance of identifying desirable antibody fragments for further therapeutic development, so these principles can be applied to any new discovery project.
INTRODUCTION
Between the time George Smith first performed the experiments to display a peptide on the surface of an M13 bacteriophage (Smith 1985) and when he was then awarded the Nobel Prize in Chemistry along with Sir Gregory Winter in 2018 for using phage display to engineer antibody-based drugs (Smith 2019), this technology became a powerful approach for the identification of peptides and antibodies with specific binding and functional characteristics. With the explosion of the development of biologic-based therapeutics in the last few decades (Lawrence 2005; Scolnik 2009; Nelson et al. 2010; Ecker et al. 2015), phage display has replaced or complemented traditional hybridoma fusion technology as a method for antibody discovery, in both academic and pharmaceutical laboratories, and the success of this approach is demonstrated by the several dozens of phage-derived antibodies in clinical development. As of 2022, 14 antibodies have been approved by the U.S. Food and Drug Administration (FDA) and/or The European Medicines Agency (EMA) for the treatment of diverse diseases (for reviews, see Alfaleh et al. 2020; André et al. 2022).
Although phage display allows for the rapid identification of panels of antibody binders to a specific target, there are several caveats to this methodology that must be considered, as well as elements of the lead identification workflow before and after phage panning that must be considered to increase the chances of success. This becomes especially important as an increasing number of academic researchers are now looking to identify therapeutics as part of their translational research programs or in collaboration with biotechnology/pharmaceutical companies. Indeed, there is growing support to encourage academic drug development (Frearson and Wyatt 2010; Everett 2015), but researchers in those settings are not necessarily trained in the process of designing and implementing an antibody discovery strategy that will yield molecules with the desirable profile to become therapeutics.
Here, we discuss some of the major considerations for developing an antibody discovery campaign with phage display that includes phage library selection, along with panning and screening approaches. These considerations typically include evaluating target characteristics, establishing antibody design goals, and creating a lead identification workflow. In addition, and as a part of this collection, we provide protocols to generate a semisynthetic library that can be used as a source of specific and developable antibodies for therapeutic development (Protocol: Semisynthetic Phage Display Library Construction: Design and Synthesis of Diversified Single-Chain Variable Fragments and Generation of Primary Libraries [Almagro and Pohl 2024a], Protocol: Semisynthetic Phage Display Library Construction: Generation of Filtered Libraries [Almagro and Pohl 2024b], and Protocol: Semisynthetic Phage Display Library Construction: Generation of Single-Chain Variable Fragment Secondary Libraries [Almagro and Pohl 2024c]).
OVERVIEW OF THE ANTIBODY DISCOVERY PROCESS
Before beginning efforts to identify a therapeutic antibody, it is important to understand the general process and workflow that must be in place for efficient selection and characterization of molecules with the desired binding and/or functional properties. Figure 1 outlines this process; each of these steps could be discussed separately in dedicated reviews, but here we simply briefly discuss each of them and focus on the use of phage display for antibody discovery. We also focus on the early stages of the process, prior to preclinical development and Investigational New Drug (IND) filing.

Development of an antibody discovery strategy. Listed are the major stages of preparation for the antibody discovery and development process, with factors for consideration at each stage to develop an antibody discovery strategy. See the text for details.
The first step in the antibody discovery process is to develop a strategy to isolate molecules with the desired functional profile that can eventually be developed into biotherapeutics. As it is often said in science, “one day of planning experiments at your desk is worth two days at the bench.” Thus, it is important to create a plan to mitigate potential risks and maximize chances for success. To begin, a molecule must be identified and validated as a relevant antigen that, when targeted with antibodies, elicits a therapeutic effect in vivo. In short, it needs to be determined whether a given molecule (1) is associated with disease, (*outlegends*f8*2) exhibits adequate expression on the cell surface or in the body fluids, and (3) is druggable by an antibody. In other words, an antibody drug is anticipated to be able to access the target in vivo and mediate a therapeutic benefit. Target identification and validation often arise from data generated in basic research laboratories. The research program may suggest, for example, that such a molecule is overexpressed in certain tumors or autoimmune disease (for reviews, see Carter et al. 2004; Hughes et al. 2011). Based on that preliminary information, a panel of proof-of-concept-specific antibodies to this molecule are generated and used for expression profiling in tumor versus normal cells, evaluating tissue specificity, or determining whether target engagement by an antibody interferes with protein–protein actions that result in alterations in signaling cascades to mediate a specific phenotype. In this case, the antibody discovery process may proceed without having fully elucidated the target biology, because the antibodies themselves will help answer these questions and thus validate the molecule as a druggable target.
The next step in developing the discovery strategy is the target feasibility assessment (Fig. 1). This process includes analysis of characteristics of the target, such as its class (e.g., soluble, membrane-bound protein, or a multipass transmembrane protein), structure, and post-translational modifications. For example, if the target is a multipass transmembrane protein, it is important to use a fragment of the protein in its native conformation as selector in the discovery campaign—and not unstructured linear peptide fragments—to identify antibodies that recognize the proper conformation of protein. Furthermore, glycosylation can significantly affect antibody binding site availability; therefore, it is prudent to determine the number of putative N-glycosylation sites on the target, which can be predicted in silico through computational tools such as PROSITE (Sigrist et al. 2013). If the target is a glycoprotein and is produced recombinantly for antibody discovery, it is important to do so in mammalian cells as opposed to in bacteria, to ensure glycosylation occurs and the molecule is in a conformation as close as possible to that of the functional native protein.
It is also important to determine the target's closest homologs in humans as well as orthologs in relevant species, mainly in rodents and nonhuman primates (NHPs), that could eventually be used for in vivo efficacy and safety studies. Knowing the level of similarity between a human target and that of its rodent and/or NHP orthologs will determine the likelihood that any antibodies identified will be cross-reactive to these species. Cross-reactivity between humans and relevant species such as rodents and NHPs used in efficacy and safety studies is often desired, so that no surrogate antibodies are needed. Moreover, if a given target is from a family of proteins with a high level of similarity among them, it may be necessary to incorporate counterscreens into the antibody screening funnel to ensure the selectivity of the antibody, and thus avoid unwanted toxicity due to targeting closely related molecules (Kushwaha et al. 2014). Alternatively, it may be wise to focus antibody generation efforts on domains of the protein that are most divergent from homologs, to improve the chances of identifying target-selective mAbs.
Once the aforementioned information on the target is obtained, a set of additional success criteria should be defined up front for the antibody discovery and optimization campaigns. These criteria include but are not limited to affinity, isotype, and potency in cell-based assays and/or in vitro functional assays, in addition to efficacy, safety, pharmacokinetics, pharmacodynamics, and developability. For instance, if the intended mechanism of action (MOA) is to disrupt a ligand–receptor interaction, then an affinity higher than the ligand's affinity to the receptor may be required. In some instances, such affinity should be in the low picomolar range (Tiwari et al. 2017). If the goal, however, requires merely binding to a membrane-bound protein, an affinity in the low nanomolar range may be sufficient. If the MOA is to block a ligand–receptor interaction, and no effector function such as antibody-dependent cellular cytotoxicity (ADCC) is desired, a human isotype such as immunoglobulin (Ig)G4 or IgG2 should be considered. On the other hand, if the MOA is supposed to destroy a tumoral cell via Fc engagement, ADCC is necessary, and thus IgG1 should be the isotype of choice.
In terms of efficacy, specific benchmarks can be set, such as, for instance, equal or greater reduction in tumor volume compared to existing therapeutics to that target in a mouse xenograft model. Depending on the intended product profile of the therapeutic, some of these criteria are more important than others. For example, if the end product is a chimeric T-cell antigen receptor (CAR-T) cell, the antibody fragment will be expressed on the patient's own cells, so manufacturability as a soluble recombinant protein is not a major consideration (Huang et al. 2020). Conversely, if the final product is an IgG, manufacturability traits such as high expression yield from the production cells, solubility in the formulation buffer, minimal or no aggregation, and short- and long-term stability are critical for development of the antibody-based drug. Alternatively, the goal may be to develop a molecule that requires less frequent dosing than current therapies. In this case, pharmacokinetics of the antibody is a top priority. Importantly, each of these antibody criteria must be as well defined as possible prior to initiation of the antibody discovery campaign and tailored to the specific antibody development program. It is essential to precisely outline as many antibody design goals as possible at the beginning of the project, as they will dictate the critical path for lead identification and optimization efforts, preclinical and clinical development costs and timelines, and, ultimately, probability of success.
The predetermined criteria defined in the antibody design goals allows for creation of a lead identification screening funnel that will be applied to any antibodies derived from a hybridoma fusion, sequence recovery from B-cell sorting, or, in our case, an antibody phage-displayed selection campaign (Fig. 2). After deciding on the selection strategy (i.e., target presentation, concentration, rounds of panning, and elution conditions, among others), the first step in most screening funnels is a primary binding assay, using either the soluble recombinant protein or cells expressing the target. From there, primary hits can be screened (or counterscreened) for cross-reactivity with orthologs from relevant species in efficacy and/or safety studies as well as with related proteins to assess selectivity. Secondary screenings can include binding to cells endogenously expressing the target of interest or evaluating binding in target knockout cells, to confirm specificity in a more relevant biological context. Triaged hits can then be produced as IgGs with the desired isotype and screened for functions such as cell-based blockage of a ligand–receptor interaction, or via a cell-based neutralization assay. It is important that in parallel to the assessment of the in vitro biological activity of those hits that have demonstrated the desired binding and functional profiles, a preliminary assessment of the developability profile of the molecules is performed, including monomeric content after purification, identity via intact mass spectrometry, thermal stability, and solubility.

General screening funnel for antibody lead identification. During a phage display-based antibody discovery campaign, polyclonal pools of binders to the target are enriched during phage panning, and then single antibody binders are identified in the primary screen. These then move to secondary screening, with select hits moving into in vivo testing and biophysical optimization and engineering. At each step, the number of antibodies is triaged, starting with hundreds or thousands of antibodies down to a few.
For those hits that have demonstrated the desired functional and developability profiles, an in vivo efficacy study can be considered. Based on the target biology, a disease-relevant animal model is selected, and parameters of the study can be established. If sufficient potency in vivo is observed as compared to either a reference molecule (if known) or a pre-established criterium (if no benchmark is available), lead candidate molecules can move into the optimization stage. At this stage, the number of hits has likely decreased from the hundreds down to a handful. Lead optimization is a field of its own, generally including a more thorough biophysical assessment of molecules, and improvement of properties such as affinity, cross-reactivity, and selectivity; expression in the manufacture platform (commonly CHO cells); stability under stress conditions; and concentration in formulation buffers. These activities usually fall outside of an academic research setting or discovery laboratory, but rather take place within specific, specialized teams in biotechnology or pharmaceutical companies.
With a clearly defined antibody discovery strategy and screening funnel, preparation of reagents and assays can begin. An essential step for the identification of a lead antibody is antigen and assay quality control (QC), which is often overlooked in academic laboratories. All recombinant proteins used for panning or screening should be assessed for purity by SDS-PAGE analysis and size exclusion chromatography. A good rule of thumb is >90% purity and visible lack of aggregation. Further QC may include testing proteins for binding to their ligands by enzyme-linked immunosorbent assay (ELISA) or kinetic methods (surface plasmon resonance, biolayer interferometry), which confirms that the antigen is in the native and relevant conformation. Additional QC can also include ELISA or kinetic assessment with a reference antibody against the target. If performing phage selection using whole cells as antigen, it is important to assess antigen density and relative expression levels of the target on the cell surface by flow cytometry.
PHAGE DISPLAY LIBRARY CONSTRUCTION AND SELECTION FOR THERAPEUTIC MONOCLONAL ANTIBODY IDENTIFICATION
Like all antibody generation methods, there are advantages and disadvantages to various types of phage display libraries and their use in a discovery campaign. There are three major types of phage display libraries, which are outlined in Table 1. Fully naive libraries are those in which the natural immune repertoire is captured, usually from healthy donors. With such a library, the repertoire has not been skewed toward one particular target, other than what the donor has naturally been exposed to in the course of their lifetime. Here, the general approach to maximize diversity is to increase the number of donors used in the library construction as well as to use primers for antibody gene amplification that fully capture the variable gene rearrangement repertoire. If careful measures are taken to amplify these gene fragments, and multiple ligations/electroporation reactions are performed, these libraries can routinely reach a very high diversity, commonly on the order of 109–1011 clones of unique antibody sequences (Almagro et al. 2019). It should be noted that a naive library is not reliant on the development of any specific immune response, and thus has the advantage that it can be used for antibody selections with a wide array of targets. This makes such a library especially attractive for therapeutic antibody discovery, as the same library can be panned repeatedly against various antigens of interest, saving significant time and cost as a laboratory moves through a portfolio of discovery programs. Well-known examples of such libraries are those developed by the Xoma Corporation, which were generated in both Fab and single-chain variable fragment (scFv) format from 30 healthy human donors (Schwimmer et al. 2013). These libraries have been panned using hundreds of targets, generating several therapeutic antibodies for clinical development, and have been licensed for use at several biotechnology companies since their inception.
Types of phage display libraries and their defining characteristics
Despite the versatility of naive libraries, antibodies from a naive library often require additional engineering, as the repertoire of antibodies did not naturally evolve before the library was generated to produce therapeutic antibodies, and thus construction of the library does not take into account important biophysical features for monoclonal antibody (mAb) development, such as thermostability, solubility at the (high) concentrations needed in therapy, and high expression yield in manufacturing platforms. Thus, naive library-derived antibodies may need to undergo affinity maturation to increase affinity or other efforts to increase stability, because this normally occurs in vivo during the B0cell maturation process, and antibodies in a naive repertoire were not all subjected to this process (Mesin et al. 2016).
Alternatively, immune libraries can be an attractive type of phage library for discovery (Table 1). These libraries have shown great value in infectious diseases such as AIDS or COVID-19 (Ubah and Palliyil 2017; Sokullu et al. 2021), where individuals are naturally infected and survive the infection by developing an immune response against the pathogen. However, in cancer or autoimmune diseases where the immunization of humans is not possible, the library is constructed using material from animals, such as mice, chickens, or llamas, immunized with the target of interest, (Lim and Chan 2016). The antibody repertoire in the immune libraries is much more focused on antibodies specific for the target used in the immunization protocol, which results in antibodies that have a higher affinity for the original immunogen, as affinity maturation occurred in vivo. Despite higher affinities, in the case where the antibodies are obtained from a nonhuman source, immune library-derived antibodies require additional downstream engineering efforts to minimize potential immunogenicity issues, a process known as antibody humanization. Pharmaceutical/biotechnology companies often perform these efforts in antibody engineering or lead optimization teams. However, the emergence of several transgenic mouse models expressing fully human immunoglobulin repertoires, such as Alloy, OmniMouse, AlivaMab, and VelocImmune mice, has allowed for the generation of human antibodies directly derived from a rodent immunization campaign, and many of these models have reached the market or are available for licensing (Murphy et al. 2014; Brüggemann et al. 2015).
A third option is the use of synthetic or semisynthetic libraries (Table 1). There is variation in the design here, ranging from the entire antibody repertoire produced artificially (fully synthetic) to a semisynthetic library where natural diversity is only introduced at particular regions of the antibody fragment; for example, at the complementarity-determining regions (CDRs) (Valadon et al. 2019; Davydova 2022). Fully synthetic libraries have predefined designed scaffold(s), with variation in certain positions of the CDRs introduced artificially by PCR or cloning of synthetic DNA fragments. To avoid introduction of stop codons in the randomized fragments that lead to nonfunctional clones, synthetic libraries such as the HuCAL Library series used trinucleotide mutagenesis (TRIM) technology to generate diversity (Knappik et al. 2000; Rothe et al. 2008; Prassler et al. 2011). Depending on the desired application of the library, the extent of the diversity either can be controlled to cast a wide net by introducing high diversity in the CDRs or can be carefully introduced at specific residues of the antigen-binding site. Synthetic libraries can also be designed with the final therapeutic product in mind, such that a particular scaffold that is known to be stable and that features desirable biophysical properties can be chosen (Tiller et al. 2013; Azevedo Reis Teixeira et al. 2021).
Semisynthetic libraries are a hybrid approach to generating a versatile library for antibody discovery. This type of library takes advantage of the control over scaffold selection and variation of select residues that is afforded by a synthetic library. However, it also introduces natural diversity in the CDRs; for instance, in the third CDR of the heavy chain (HCDR3), which is commonly (but not always) the most important region of the binding site for antigen interaction and is difficult to design due to the high diversity and flexibility of this region (Almagro et al. 2014; D'Angelo et al. 2018). Examples of semisynthetic libraries, which are described in detail below and for which protocols for construction are included in this collection, are ALTHEA Gold Plus Libraries (see Protocol: Semisynthetic Phage Display Library Construction: Design and Synthesis of Diversified Single-Chain Variable Fragments and Generation of Primary Libraries [Almagro and Pohl 2024a], Protocol: Semisynthetic Phage Display Library Construction: Generation of Filtered Libraries [Almagro and Pohl 2024b], and Protocol: Semisynthetic Phage Display Library Construction: Generation of Single-Chain Variable Fragment Secondary Libraries [Almagro and Pohl 2024c]).
Like naive libraries, synthetic and semisynthetic libraries can be valuable assets for a laboratory focused on antibody discovery, as they are target-agnostic and can be used to isolate antibodies for virtually any target of interest. Moreover, these types of libraries allow for the identification of antibody binders without dependence on a lengthy immunization protocol and a robust immune response. However, although it has been reported that high-affinity binders can be identified from synthetic (Ferrara et al. 2022) and semisynthetic libraries (Guzmán-Bringas et al. 2023), some antibodies will require additional engineering to achieve the desired affinity.
Besides the source of the antibody repertoire to be used in a naive, synthetic, or semisynthetic library, it is also important to select features of the library that are conducive to the discovery workflow of interest and desired output. This includes the format of the antibody fragment that will best fit the therapeutic modality in development. For example, if the desired final molecule will be a conventional IgG, a Fab phage display library may be preferable, as it is closer to the final format of the therapeutic. Fabs are commonly more stable than scFvs, as they are stabilized by the CH1:CL interface and a C-terminal disulfide bridge in addition to the VH:VL interface of the scFvs. However, Fab libraries can be more difficult to construct and may not be as well expressed in Escherichia coli as a smaller scFv fragment (Omar and Lim 2018). Efforts have been made, however, to optimize scFv construction (Zhu and Dimitrov 2009).
Other features of the library must also be carefully selected. For scFv libraries, selection of the proper format, either VH–linker–VL or VL–linker–VH, and the nature of the linker (usually a flexible glycine–serine repeat) between the VH and VL fragments can impact the outcome of the selection process. Longer linkers allow for the scFv to fold back on itself, promoting monomer formation, whereas shorter linkers lead to dimerization between VH and VL of two scFvs, thus forming diabodies (Arndt et al. 1998). Diabodies could result in apparent affinity increases due to multiple antibody fragments interacting with the target, often referred to as avidity effect (Rudnick and Adams 2009), which departs from the true affinity of the IgG that is the most common final therapeutic format of antibodies. Some other standard features of a phagemid vector include the presence of an amber stop codon to allow for soluble antibody production free of the pIII fragment directly from the phagemid vector, as well as a tag for detection. Oftentimes, 6X-His, myc, HA, or V5 tags are chosen, but depending on what tags are commonly found on the antigens you will be panning against, you may want to avoid one or more of these. For instance, the His tag is very useful for scFv purification via nickel or cobalt affinity columns as well as in detection assays using anti-His antibodies. However, many commercially available antigens have His tags, and thus a His-tagged scFv may be a liability to developing assays with anti-His detection antibodies.
A CASE STUDY IN LIBRARY CONSTRUCTION FOR THERAPEUTIC MONOCLONAL ANTIBODY DISCOVERY: THE ALTHEA GOLD PLUS LIBRARIES
ALTHEA Gold Plus Libraries are an upgrade of ALTHEA Gold Libraries, described in detail by Valadon et al. (2019). A comparison of ALTHEA Gold Libraries with state-of-the-art antibody phage-displayed libraries described in the literature over the last 10 yr has been discussed by Almagro et al. (2019). Among others, these libraries include naive libraries implemented by Xoma (Schwimmer et al. 2013) and synthetic libraries such as HuCAL Platinum (Prassler et al. 2011) and Ylanthia (Tiller et al. 2013) implemented by Morphosis, which are used by large pharmaceutical and biotechnology companies as their antibody discovery platforms.
ALTHEA Gold Libraries consist of two scFv sublibraries built with synthetically generated, well-known human IGHV and IGKV germline genes, combined with natural human HCDR3/JH (H3J) fragments obtained from peripheral blood mononuclear cells from a large pool of healthy human donors. One synthetic IGHV gene (IGHV3-23) provides a universal variable domain of heavy chain (VH) scaffold, paired with two variable light (VL) scaffolds obtained from the IGKV3-20 and IGKV4-01 genes that furnish two different topographies at the antigen-binding site, and hence the potential to bind distinct epitopes on the target.
All CDRs in these libraries are synthetically diversified, with the exception of the HCDR3. A strategic approach was implemented for synthetic VH and VL generation, whereby scaffolds are mutated at specific positions identified as being in contact with antigens in the known antigen–antibody complex structures that were available at the time of the library design (more than 2000 antibody structures are available at the Protein Data Bank, https://www.rcsb.org/). The diversification regime consists of high-usage amino acids found at those positions in corresponding human germline genes, and more than 60,000 antibody sequences compiled at the National Center for Biotechnology Information (https://www.ncbi.nlm.nih.gov/) and the international ImMunoGeneTics information system (https://www.imgt.org).
In addition to the unique design of the ALTHEA Gold Libraries, the functionality, stability, and diversity of the libraries are improved throughout a three-step construction process (Fig. 3). In the first step, fully synthetic primary libraries (PLs) are generated by combining the diversified scaffolds with a set of 90 synthetic neutral H3J germline gene fragments. These neutral H3J fragments serve as a “placeholder” to avoid structural constraints that a single H3J fragment could impart during subsequent steps in the library construction process and result in limited sequence diversity in the final libraries.
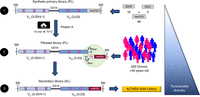
Three-step construction process to generate highly diverse and functional semisynthetic libraries. Figure reproduced from Valadon et al. (2019). See the text for details.
The second construction step consists of selecting or filtering PLs, based on the natural capacity of the Staphylococcus aureus protein A or of the Peptostreptococcus magnus protein L to bind human VH or VL scaffolds, respectively. Protein A binds the framework 3 of human IGVH3 gene family members (Graille et al. 2000), whereas protein L binds the framework 1 of human IGKV1, IGKV3, and IGKV4 gene families (Nilson et al. 1992; Graille et al 2002). These properties of protein A and protein L have been extensively used to select for well-folded and stable antibody fragments after incubation under denaturing or destabilizing conditions (Jespers et al. 2004; Hussack et al. 2011). However, the use of protein A or L has been limited to enriching the final antibody libraries and/or selecting for variants after mutagenesis to improve stability. In the accompanying protocols, we use protein L to generate filtrated libraries (FLs) as an intermediary step to enrich the PLs with well-folded antibody fragments, taking place after harsh incubation conditions and before replacing the neutral H3J fragments with natural H3J fragments (see Protocol: Semisynthetic Phage Display Library Construction: Design and Synthesis of Diversified Single-Chain Variable Fragments and Generation of Primary Libraries [Almagro and Pohl 2024a]; Protocol: Semisynthetic Phage Display Library Construction: Generation of Filtered Libraries [Almagro and Pohl 2024b]; Protocol: Semisynthetic Phage Display Library Construction: Generation of Single-Chain Variable Fragment Secondary Libraries [Almagro and Pohl 2024c]). In the filtration process, the well-folded antibody fragments fused to phage particles bind to protein L, whereas truncated fragments resulting from premature stop codons or nonfunctional ones resulting from insertions and/or deletions are removed by centrifugation. These nonfunctional antibody variants erode the repertoire of functional antibody fragments in the library, leading to poor performance, so it is important that they are removed.
In the third and final step, the resulting stable synthetic antibody fragments of the FLs are combined with natural H3J fragments to generate secondary libraries. Because most of the antibody diversity is concentrated in the HCDR3, the natural H3J fragments restore the diversity lost during the filtration process. Moreover, because the natural H3J fragments have been under selective pressures to produce functional antibodies in vivo, the combination of highly stable synthetic antibody fragments from the FLs and a repertoire of natural H3J fragments results in highly diverse, stable, and functional libraries ready for therapeutic antibody discovery.
Validation of ALTHEA Gold Libraries with seven targets yielded specific antibodies in all cases. Further characterization of the isolated antibodies indicates KD values as human IgG1 molecules in the single-digit and subnanomolar range. Thermal stability (Tm) of all tested Fabs was between 75°C and 80°C, demonstrating that ALTHEA Gold Libraries are a valuable source of specific, high-affinity, and highly stable antibodies. Several target-specific antibodies isolated from these libraries have been characterized in detail (Pedraza-Escalona et al. 2021; Dao et al. 2022a,b), with one mAb even moving to Phase 1 clinical trials (NCT06017258 and NCT06128044).
The upgrade to ALTHEA Gold Plus Libraries from ALTHEA Gold Libraries results from adding two additional VL scaffolds (IGKV1-39 and IGKV3-11), for a total of four scFv semisynthetic libraries. These new scaffolds increase the structural diversity of the libraries, with the potential to generate a more diverse set of antibodies. Recently, application and validation of the four synthetic VL scaffolds used in ALTHEA Gold Plus Libraries were published by Mendoza-Salazar et al. (2022). In this work, the four synthetic VL libraries were used as counterparts of an immune VH repertoire obtained from a patient with coronavirus disease 2019 (COVID-19) infected with the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) B.1.617.2 (Delta) variant. Using standard molecular cloning techniques, the variable light chain diversity of the ALTHEA libraries was combined with the VH repertoire from the COVID-19 convalescent patient. After three rounds of selection with SARS-CoV-2 wild-type (WT) receptor-binding domain, 34 unique scFv molecules were obtained. One of the antibodies, called IgG-A7, recognized the SARS-CoV-2 receptor-binding domain from Wuhan-Hu-1 (WT), Delta, and B.1.1.529 (Omicron) strains with high affinity. IgG-A7 also neutralizes the three strains of SARS-CoV-2 in plaque reduction neutralization tests and protects 100% of K18-hACE2 transgenic mice expressing human angiotensin-converting enzyme 2 (hACE2) infected with WT SARS-CoV-2 at a dose of 5 mg/kg (González-González et al. 2022). The developability profile of IgG-A7 matches that of marketed therapeutic antibodies, thus demonstrating the potential of the synthetic VL scaffolds used in ALTHEA Gold Plus Libraries to generate high-affinity and functional antibodies with potential as leads for development of antibody-based drugs.
The three accompanying protocols for implementing ALTHEA Gold Plus Libraries include Protocol: Semisynthetic Phage Display Library Construction: Design and Synthesis of Diversified Single-Chain Variable Fragments and Generation of Primary Libraries (Almagro and Pohl 2024a), Protocol: Semisynthetic Phage Display Library Construction: Generation of Filtered Libraries (Almagro and Pohl 2024b), and Protocol: Semisynthetic Phage Display Library Construction: Generation of Single-Chain Variable Fragment Secondary Libraries (Almagro and Pohl 2024c), and below we discuss general considerations to increase the chances of success during the implementation of the protocols.
DEVELOPING A PANNING STRATEGY
After following the elements discussed above, a library that fits the needs of your discovery strategies can be generated, and a discovery campaign can be started. The first step in this process is to develop a strategy based on the characteristics of the target and available reagents. For instance, the nature of the target will have an important impact on the antibody design goals. If the target or a relevant fragment of the target can be produced as a high-quality recombinant protein that passes QC analysis, this provides a straightforward approach for the identification of binders. The simplest method of phage panning with soluble protein is a solid phase panning, whereby the target antigen is immobilized on a polystyrene surface, on either a microwell plate or immunotube (Kontermann 2001; Russo et al. 2018). This simple panning approach, however, has the disadvantage that protein adherence to the plastic may cause minor structural changes, resulting in the protein not having the same conformation as in solution. This may affect the conformation or availability of functional epitopes important for obtaining antibodies with the desired functional profile (Carmen and Jermutus 2002). In addition, in solid phase panning, the amount of antigen attached to the polystyrene cannot be controlled as well as in solution, as only a fraction of the protein added to the well/tube will adhere to the plate and saturate the available surface.
Alternatively, panning can be performed in solution, using the soluble target with an affinity tag for capture/immobilization, often biotin (Wenzel et al. 2020). In this panning modality, the antigen–biotin conjugate can be isolated using magnetic nanobeads coated with streptavidin, and the nonbound phage in solution can be separated from the bound phage. The twofold advantage of this process is that (1) the concentration of the protein captured by the beads (and the number of beads) can be controlled, which is especially important when trying to select for higher-affinity antibodies, and (2) the target is in its native conformation in solution, thus avoiding conformational changes that may occur in solid phase panning. One potential issue with this panning method, however, is that because the antigen requires labeling, the biotin conjugation could result in blocking desired epitopes. This can generally be avoided, though, by introducing a site-specific biotinylation tag on the antigen expression construct, such as an AviTag, so that the degree of labeling is 1:1 and the biotinylation occurs in a region that does not compromise the integrity of functional epitopes (Gräslund et al. 2017). Some commercial vendors offer Avi-tagged proteins for this purpose.
Some of the most challenging targets to isolate antibodies against include MHC complexes loaded with specific peptides to generate TCR-mimic antibodies, membrane-bound proteins, and ion channels. In the case of MHC:peptide complexes, there are challenges in generating appropriate antigens for selection and confirming that antibodies identified from the panning campaign are specific for the MHC in complex with the particular peptide of interest. One approach here is the use of biotinylated peptide–MHC antigens, and established methods for producing such complexes in bacteria have been published (Denkberg et al. 2000). Two case studies of identification of highly specific antibodies to peptide–MHC complexes by phage display are described by Dao et al. (2022a,b).
Membrane proteins, on the other hand, are difficult to express in soluble form, and the exposed exterior surface of the proteins in some cases are peptides that conformationally depend on the protein embedded in the membrane, and thus are difficult to produce as recombinant functional protein fragments. Ion channels are similarly notoriously difficult targets to generate antibodies against (Wilkinson et al. 2015), as often the goal is to isolate an antibody that blocks function due to occlusion of the channel pore by the antibody. This often leaves little “real estate” for an antibody to bind to, and is reliant on the pore complex to be present in its native, multimeric complex, ideally in one conformation (pore open or closed). In this case, whole cells or membrane fractions can be used for panning, or rather virus-like particles (VLPs), nanodisks, or amphipols loaded with the ion channel of interest. Amphipols consist of an amphiphilic polymer designed to serve as a “belt” to constrain the oligomer into its native membrane-bound conformation, resulting in a soluble antigen that can be used for panning (Hou et al. 2020). Similarly, nanodiscs are disc-shaped complexes consisting of a lipid bilayer surrounded by amphipathic protein that can also constrain a membrane protein suitable for phage selections (Pavlidou et al. 2013). Specificity can be an issue, however, as both the extracellular and intracellular portions of the ion channel are exposed in the phage panning mixture. For a review of antibodies generated against ion channels, see Haustrate et al. (2019).
Another approach to isolate antibodies against difficult targets using phage display is cell-based panning (Fahr and Frenzel 2018). In this panning modality, the phage library is directly incubated with cells expressing the target of interest. The advantage of this method is that the target of interest is in its native conformation and functional configuration. One of the drawbacks of cell-based panning, however, is that the expression level of the target may not be sufficient, and so it may be difficult to isolate binders to the protein. Another major difficulty with cell-based panning is specificity, as there are many other proteins than the target of interest on the cell surface that will compete for binding to the antibodies in the library, and thus can dampen the enrichment of specific binders to the target. Thus, it is important to first deplete the phage library of nonspecific binders and binders specific for proteins other than the target of interest with a cell line that does not express the target of interest. This step, called subtraction or negative selection, is ideally performed with the cell line used to generate the recombinant cell line; i.e., the cell line that has not been transfected with the target. Alternatively, one can alternate between soluble antigen and cells expressing the target to selectively isolate the antibodies binding both and remove phages that bind to irrelevant targets on the cell surface.
OPERATIONAL CONSIDERATIONS BEFORE STARTING A PHAGE PANNING CAMPAIGN
Once a phage display library has been constructed, it is important to store the library under conditions to maintain its diversity. A library can be stored either as purified virions, or as a frozen bacterial stock that requires culture growth and infection with helper phage for amplification of virions displaying antibodies. If a library is stored as phage stock, it should be mixed with glycerol at 50% (v/v) and stored at −80°C, and should not be subjected to multiple freeze–thaw cycles. It should be noted that this is not to protect the phage virions themselves, which are quite robust and stable (Branston et al. 2013), but rather to minimize irreversible unfolding of the antibody fragments expressed on the phage surface and/or degradation by traces of proteolytic enzymes present derived from the originating bacterial culture. Both processes will lead to significant losses in diversity of the library when panning, but are not reflected in the titer of the phage library, as the phage particles are still infectious regardless of the presence of a displayed antibody. To avoid losses in library diversity due to antibody fragments unfolding and degradation, it is common practice to avoid large phage stock preparations and use freshly amplified libraries from bacterial stock as input for the first round of panning when possible. Of course, library preparation every time a new campaign is started is time- and resource-consuming, and thus a proper balance must be struck between having a phage stock ready and preparation right before a discovery campaign, depending on the demands of a given laboratory. It is important to note that it is never ideal to amplify a library from an existing phage stock, as this practice introduces amplification bias of certain clones as an unintended consequence of growth advantage of some clones (particularly phage with defective antibody fragments or without the fusion proteins displayed altogether) over others that may express valuable antibodies, which can adversely affect library diversity (Matochko et al. 2012).
Phage display panning campaigns require relatively standard equipment and reagents that are usually found in laboratories running routine experiments in molecular biology and biochemistry. With that said, there are several steps when preparing to efficiently execute rounds of phage panning. This is akin to the “mise-en-place” philosophy of chefs, which translates to “everything in its place.” The idea is to have all ingredients on hand and prepared before you start cooking or, in this case, phage panning. It is important to ensure that all instruments are in working order, e.g., centrifuges, bacterial shaking incubators, spectrophotometer, among others, and that all reagents, including high-quality antigen, helper phage stock, culture media, and buffers, are ready and in sufficient supply. The laboratory bench and pipettes should be decontaminated with 10% (v/v) bleach, especially if multiple panning campaigns are to be performed in parallel, and sterile plastic disposable material is preferred over reusables.
Time management of panning rounds is another important consideration. Depending on the protocol, a single round of panning can be completed in 1 d, or it can be broken up into 2 d. In the 1-d scenario, the panning is completed, and eluted phage is amplified in a bacterial culture overnight, to be harvested fresh the next day for the subsequent round of panning. On this schedule, a panning campaign can be completed in 1 wk. However, one runs the risk of having to start over from the beginning (Round 1) if anything happens during later rounds of panning to the eluate or amplified eluate, as there is no “backup” stock. Alternatively, in the 2-d method, a bacterial culture is infected with eluted phage and spread onto solid medium for overnight growth. Scrapings from the agar plate can then be used the next day to grow up a log phase bacterial culture for helper phage infection and phage production. The advantage here is threefold: (1) Plating the eluate onto solid media allows for more slow-growing clones to “catch up” to those that would have a growth advantage in liquid medium, potentially increasing diversity of the eluted phage pool; (2) one can in parallel titer the output of the round of panning or just see the density of the bacteria, and thus assess how the panning is going; and (3) glycerol stock of the plate scrapings can be stored to preserve each round of panning. The bacterial glycerol stocks can be used to reamplify phage from that round for repeating panning, panning on a different antigen, or isolation of single colonies from that round for monoclonal screening.
It is also crucial to avoid contamination while panning, especially with helper phages. Due to the infectious properties of helper phages, phagemids, and F-pilus-expressing E. coli (Boeke et al. 1982), if the bacterial culture to be used for eluate amplification first becomes infected with helper phages, it deems those bacterial cells impervious to infection with phagemid virus, which can result in a loss of a panning round. Also, if performing more than one campaign in parallel, it is important to avoid cross-contamination. The best way to avoid contamination is to follow good laboratory practice, including the use of filter pipette tips and disposable plastic cultureware, and disinfecting surfaces frequently with freshly prepared 10% (v/v) bleach.
TECHNICAL CONSIDERATIONS TO ISOLATE ANTIBODIES OF INTEREST
Regardless of the panning approach used, i.e., solid phase, solution panning, or cell-based selection, it is important to incorporate the overall antibody design goals into the panning schema. For example, if cross-reactivity to mouse or NHP orthologs of the target is required, these targets can be incorporated into alternating rounds of selection to enrich for binders that recognize shared epitopes between species. In contrast, if selectivity is required, and there are specific isoforms, paralogs, or related proteins in the target protein family, a depletion or subtraction step can be added at the beginning of each panning round, to remove cross-reactive binders with the unwanted targets from the phage library.
Besides the target selection, additional measures can be taken to isolate antibodies of interest. For example, if antibodies with higher affinity are desired, the target concentration can be decreased successively over each round of panning, usually fivefold to 10-fold per round. The easiest way to accomplish this is in a solution panning, where the target concentration can be tightly controlled. As the phage pool is enriched for binders to the target over each round of panning, lowering the antigen concentration creates competition in solution, with the highest-affinity antibodies retaining binding and outcompeting lower-affinity binders for antigen engagement.
Another way to select for higher-affinity antibodies is to increase washing stringency over successive panning rounds; for example, 10 washes in Round 1, 15 washes in Round 2, and 20 washes in Round 3. Adding detergent such as Tween to the wash buffer can help decrease nonspecific interactions between phage and the target, thus helping to isolate antibodies of interest. The washing stringency can also be increased by having longer incubation times in the wash buffer. This will select for antibodies with a slower off rate, and thus remove binders with a lower dissociation constant. In phage-based affinity maturation campaigns, wash times will often be extended to 2 h and even overnight to select for the highest-affinity clones (Thie et al. 2009). Additionally, one can add an excess of nonbiotinylated target during the overnight incubation, to outcompete the lower-affinity binders bound to the target. Finally, it should be considered that the elution method can affect the specificity of clones isolated. Instead of a general, nonspecific elution method using extreme pH such as Tris-glycine pH 2.2 or triethylamine pH 10, which will indiscriminately disrupt antigen–antibody interactions, a more focused competitive elution can be used. In this elution strategy, a nonbiotinylated target or a known functional antibody can be added to the phage pool to compete for phage-displayed antibodies. With this approach, only target or epitope-specific clones will be eluted from the phage–bead complexes (Duan and Siegumfeldt 2010).
Although affinity is important for many therapeutic antibody programs, in some cases it is not as critical as obtaining antibodies binding the right epitope to achieve the desired function. Indeed, selecting a diverse panel of antibodies is often more important than obtaining a few high-affinity binders. The kinetics of a functional antibody can be improved via an affinity maturation campaign, but a high-affinity antibody cannot be engineered into a functional antibody it if does not bind the right epitope. Thus, the initial goal of a discovery campaign should be focused on identifying antibodies that recognize as many epitopes as possible, to increase the chances of targeting the right functional epitope, so it can be used for proof-of-concept studies or as substrate for further optimization. If this is the case, panning selection conditions can be tweaked accordingly. To isolate a more diverse pool of antibodies, the selection conditions in the first round of panning are most important, because these are the clones that will be carried throughout the rest of the selection rounds. If conditions are too stringent in the first round, diversity will be significantly compromised. To avoid this pitfall, a higher concentration of antigen and a minimal number of washes can be used in Round 1, to avoid discarding lower-affinity but potentially functional binders. Also, fewer rounds of panning can be performed to avoid any clonal selection/outgrowth in later panning rounds. Keep in mind, however, that the diversity of the antibodies isolated from any campaign is limited by the source library, which is why careful construction of highly diverse and functional libraries is critical for a successful discovery campaign.
CONCLUSION
There are many variables in a therapeutic antibody discovery campaign that must be considered before and during the lead identification process to increase the chances of success. With that said, once streamlined standard protocols, processes, and reagents are established in an antibody discovery laboratory (e.g., protein/antibody production and QC capabilities, a validated naive phage display library or immune library construction protocol, and high-throughput screening methods), campaigns can run efficiently for rapid identification of antibody candidates for further characterization. The most crucial portions of this workflow are (1) the antibody phage display library as the source of binders, as it will have the biggest impact on sequence diversity and quality of recovered antibodies; (2) the quality of the reagents, in particular the targets, as they will determine whether one is selecting for the right conformation/function; and (3) validated assays that will guide the decision on the best clones to move forward in the discovery and optimization campaigns. Moreover, the basic steps during the selection process, such as binding, washing, elution, and phage amplification, generally remain consistent among discovery campaigns, but minor changes in the selection protocol can significantly affect the results of the campaign, so careful recordkeeping of experimental variations is critical. Using information from previous antibody discovery efforts can also help to move a program forward. Ultimately, however, the success of a campaign is often determined empirically. It is best to try both proven and new creative panning approaches to deliver a diverse panel of antibody sequences for further testing, as hundreds of antibodies will eventually be narrowed down to a few leads for full preclinical development efforts.
In summary, the following points will pave the way for a successful phage-based lead identification approach. First, establish antibody design goals. Second, library source and method of construction have the biggest effect on sequence diversity and utility, so carefully select or build a library that will best fit the antibody design goals of the project. Third, perform rigorous quality control assessment of all reagents. Finally, design and implement library selection strategies and screening methods that maximize the chances of enrichment for antibodies with the desired target binding and functional properties.
COMPETING INTEREST STATEMENT
M.A.P. is an employee of Xtalpi, Inc. J.C.A. is founder and CEO of GlobalBio, Inc., and has commercial interest in ALTHEA Gold Plus Libraries.
Footnotes
-
From the Advances in Phage Display collection, edited by Gregg J. Silverman, Christoph Rader, and Sachdev S. Sidhu








