
Organ Culture and Immunostaining of Mouse Embryonic Kidneys
- Department of Developmental Biology, Washington University School of Medicine, St. Louis, Missouri 63110, USA
- ↵2Corresponding author (boyles{at}wustl.edu).
INTRODUCTION
The study of organogenesis in mammals allows investigation of a wide variety of basic cell biological processes in the context of the intact organ. This has become especially important in the age of genetics, as the consequences of gene deletion or mutation in the mouse can be directly linked to human congenital abnormalities. The ability to culture some organs ex vivo during development has emerged as an important tool to understand how tissues are constructed and the signaling pathways that regulate these processes. It has been especially useful in organs that grow via branching morphogenic mechanisms, such as the lung and kidney. Here we demonstrate isolation, ex vivo growth, and fluorescent immunostaining of mouse embryonic day 12.5 (E12.5) kidneys. To demonstrate nephron formation using live imaging, we have isolated and cultured kidneys from mice carrying a green fluorescent protein (GFP) transgene driven by the Hes1 promoter, which is expressed early in the developing nephron. We also provide a protocol for robust imaging of multiple kidney structures in the whole-mount setting. These techniques serve as a basic platform for the analysis of branching morphogenesis and nephron formation in genetic mouse models or in response to exogenous factors, such as agonists or inhibitors, which can be directly added to the culture medium.
RELATED INFORMATION
For previous examples and variations of this method, see Giuliani et al. (2008), Gupta et al. (2003), and Saxen (1987). For detailed reviews of the structural and genetic features of kidney development, see Costantini and Kopan (2010), Dressler (2009), and Vize et al. (2003).
MATERIALS
Reagents
Antibodies, primary (diluted to final concentrations in PBS-BB)
In our experience, kidneys are amenable to a wide variety of primary antibodies. Optimal dilutions should be determined empirically. The following examples are used in this protocol:
-
Rat anti-Cytokeratin 8 (TROMA-I; 1:50 dilution; supernatant) (Developmental Studies Hybridoma Bank [DSHB])
-
Rabbit anti-Cadherin 6 (1:400 dilution ) (Cho et al. 1998)
-
Biotinylated Lotus Tetragonolobus Lectin (LTL; 1:200 dilution) (Vector Laboratories B-1325)
-
Mouse anti-WT1 (1:100 dilution) (Dako M3561)
Antibodies, secondary (diluted to final concentrations in PBS-BB)
The following examples are used in this protocol:
-
Donkey anti-Rat Dylight 649 (1:200 dilution, far red) (Jackson ImmunoResearch 712-495-153)
-
Goat anti-Rabbit Alexa Fluor 488 (1:500 dilution, green) (Invitrogen/Molecular Probes A-11070)
-
Donkey anti-Mouse Cy3 (1:500 dilution, red) (Jackson Immunoresearch 715-165-150)
-
AMCA Avidin D (1:200 dilution, blue) (Vector Laboratories A-2008)
Ethanol (70%)
Mouse, pregnant
In the mouse, kidneys can be isolated and cultured effectively from E11.5 to E13.5. In this example, kidneys are isolated at E12.5.
Paraformaldehyde (4% in PBS; Fisher Scientific O4042-500)
Prepare fresh before use and keep cold.
PBS, cold (Invitrogen/GIBCO 14190)
PBS containing 0.1% Tween 20 (Fisher Scientific BP337-500; PBST)
Serum-free medium for kidney culture
Vectashield mounting medium
Equipment
Dishes, six- and 24-well (TPP 92006, 92424)
Filters, transwell (cell culture inserts for six-well plates, 1.0-μm pores, transparent PET membrane) (BD Falcon 353102)
Forceps, Dumont #5 (Fine Science Tools 11295-00)
Ice
Incubator preset to 37°C, 5% CO2, humidified
Laminar flow hood
Metal support grids, stainless steel
Make the grids from aluminum patio screen. Cut the screen to the size of the center well of the culture dish (Fig. 1A,B), and use a standard paper hole-punch to create a hole in the middle of the grid. Sterilize the grids before each experiment by soaking them in 70% ethanol, drying, and autoclaving. Before using the screen in an experiment, test it to ensure that the culture medium does not corrode the metal.

Dish setup for cultured kidneys. (A,B) Top and (C) side views of complete center-well dishes. Metal mesh is cut to fit inside of the center well, on top of culture medium. Filters are placed over the hole in the center of the mesh with medium contacting, but not covering, the filter (A, dashed line). (D) Filter after 48 h of culture. Up to five kidneys (black arrow) can be comfortably spaced on the filter.
Micropipettor (200 μL) and tips (wide-bore, or standard tips with the end cut off)
Microscope, dissecting/live imaging (e.g., Leica MZ10F)
Microscope, imaging (e.g., Axio Imager.Z1, equipped with Zeiss ApoTome; Zeiss)
Organ culture dish, 60-mm with a 3-cm2 center well (BD Falcon 353037)
Pad, absorbent (for dissection)
Petri dish, 60 mm × 15 mm (BD Falcon 353002)
Platforms, rocking (4°C and room temperature)
Refrigerators or cold room preset to 4°C
Scalpel and blades, #11 (Fine Science Tools 10011-00)
Slides and coverslips
METHOD
To prevent contamination, all solutions, tools, and equipment should be sterile. Prepare medium and plates in a laminar flow hood. We also recommend performing the embryo dissection in a laminar flow hood equipped with a dissecting microscope. This precaution is not required, but it greatly reduces the risk of contamination.
Preparation of the Organ Culture Dish
-
1. Fill the center well of an organ culture dish (60-mm dish with a 3-cm2 center well) with serum-free medium for kidney culture (~1.5 mL).
-
2. Use forceps to place a metal support grid in the center well.
The grid should be flat and comfortably supported by the well (Fig. 1A-C). The medium should contact the grid; if it does not, add enough medium so that it does.
-
3. Remove the filter from a transwell insert by cutting around the outside edge with a sterile scalpel.
-
4. Place the filter in the top of a 60-mm Petri dish and cut it into quarters.
-
5. Place a filter quarter over the hole in the center of the grid.
The medium should contact the filter with no air bubble underneath. If placed correctly, the filter will be held in place by surface tension (see Fig. 1A,B).
-
6. Place the culture dish in a fully humidified 37°C incubator with 5% CO2.
Isolation of Metanephric Kidneys from Embryonic Mice
Procedures for the humane treatment of animals must be observed at all times. Consult your local animal facility for guidelines. The techniques performed in this study were approved by the Washington University Division of Comparative Medicine (Protocol 20080020).
-
7. Euthanize the pregnant mouse by cervical dislocation.
See Troubleshooting.
-
8. Lay the mouse on an absorbent pad and wash the abdomen with 70% ethanol to reduce the risk of contamination (Fig. 2A).
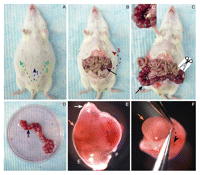 FIGURE 2.
FIGURE 2.Harvest of uterus and embryos from pregnant mice. (A-D) Removal of uterus from female. (A,B) Location and direction of cuts for opening the abdominal cavity (arrows). (Black arrows) The uterine horn; (AC) abdominal cavity. (C, yellow dashed line) Cuts to remove the uterine horn. (C') Higher magnification of the uterine horn; (yellow arrow) the location of the mesometrium. (D) Isolated uterine horn. Cut between each sac (blue arrow) to separate individual embryos. (E) Individual embryo (E) with placenta (P) in uterus; (white arrow) location where forceps are inserted to remove embryo. (F) Removal of embryo from the uterus. Forceps are used to tear muscle along the black dotted arrow to release the embryo; (orange arrows) the location of the head.
-
9. Lift the skin with forceps and make an incision at the midline, cutting through the viscera and the peritoneum, into the abdominal cavity (Fig. 2A, blue arrow).
-
10. Make two additional diagonal cuts away from the midline (Fig. 2A, green arrows). Expose the abdominal cavity by lifting the skin up and away from you (Fig. 2B, red arrow).
-
11. Dissect the uterine horn.
-
i. Grasp the uterine horn (found underneath the intestine) (Fig. 2B,C, black arrow) and separate it from the other organs in the abdominal cavity.
-
ii. Remove the uterine horn by holding it at one end and cutting along the mesometrium (Fig. 2C′, yellow arrow) and the blood vessel connection on the dorsal side of the uterus (Fig. 2C, dashed yellow line).
-
-
12. Place the dissected uterine horn in a Petri dish on ice containing cold PBS (Fig. 2D).
-
13. Separate the embryos from the uterus.
-
i. Cut the uterus between each embryo (e.g., see Fig. 2D, blue arrow).
-
ii. Transfer the Petri dish to a dissecting microscope.
-
iii. Insert forceps at the end of each section of uterus to create an opening (Fig. 2E, white arrow).
-
iv. Tear the uterine muscle with the forceps along this line to release the yolk sac (containing the embryo) and the placenta (Fig. 2F, dashed lines; Fig. 3A).
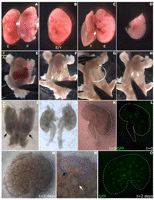 FIGURE 3.
FIGURE 3.Isolation and live imaging of kidneys from E12.5 embryos. (A-C) Separation of embryo proper from placenta and yolk sac. (E) Embryo; (P) placenta; (Y) yolk sac; (arrow) location of umbilical cord. (D) Removal of the head aids dissection and is a convenient source of tissue for genotyping. (E-H) Isolation of urogenital system. (A) Anterior; (P) posterior. The embryo is pinned with forceps at the level of the heart, and superficial organs are removed so that the urogenital system (F, dashed lines) is visible. Arrows indicate anterior end of nephric ducts, where “peeling” (G, curved arrow) is initiated. Kidneys will appear at the posterior end of the urogenital system, at the level of the hindlimb (H, arrows). (I,J) Kidneys (arrows in I) are separated from the urogenital system. (K,O) Live imaging of isolated kidneys at the beginning of the organ culture (t = 0; K,L) and after 2 d of culture (t = 2 d; M-O). Brightfield images (K,M,N) demonstrate growth of cultures. Developmental structures such as the metanephric mesenchyme (blue dotted line), ureteric bud (red dotted line), renal vesicle (black arrow), and S-shaped body (white arrow) are easily identified at higher magnifications (N). Live GFP from the Hes1-GFP transgene (L,O) labels the S-shaped body of the forming nephrons.
-
-
14. Isolate the embryos.
-
15. Move each embryo to an individual 60-mm Petri dish containing 4-5 mL of cold PBS.
-
16. Remove the head of the embryo above the forelimb (Fig. 3D). This tissue can be collected and used for polymerase chain reaction (PCR) genotyping.
-
17. Remove superficial organs.
-
i. Position the embryo with its dorsal side down (back against the plate) and posterior away from you as flat as possible.
Removal of the tail often helps to prevent the embryo from rolling (Fig. 3E).
-
ii. Pin the embryo to the plate with forceps at the level of the heart.
-
iii. Cut the embryo along the midline using forceps or the dull side of a #11 scalpel blade (Fig. 3E).
-
iv. Using the scalpel or forceps, remove the heart, lungs, liver, and intestine (Fig. 3F).
See Troubleshooting.
-
-
18. Isolate the urogenital system.
At this stage, the metanephric kidneys are located anterior to the cloaca in the lower abdominal cavity at the level of the hindlimbs, and are not readily visible. The most effective way to isolate them without damage is to remove the entire urogenital system (Fig. 3F, outline).
-
i. Identify the anterior end of the nephric ducts (Fig. 3F, arrows).
-
ii. Using the dull side of the scalpel blade or forceps, lift this sheet of tissue and carefully peel back toward the cloaca (Fig. 3G).
-
iii. Once the kidneys are visible (Fig. 3H, arrows), apply sufficient pressure to separate the urogenital system from the rest of the embryo.
If done properly, the urogenital system should all come out as one piece (Fig. 3I). When removing it in this manner, you will flip the orientation of the kidneys. Whereas before, posterior was away from you (Fig. 3E), it is now toward you (Fig. 3I).
See Troubleshooting.
-
-
19. Isolate the kidneys.
The metanephric kidneys can be detected at the posterior edge of the ventral side of the removed block of tissue (Fig. 3I, arrows).
-
i. Using forceps, carefully separate each kidney from the surrounding tissue (Fig. 3J).
-
ii. If necessary, use a scalpel to separate the kidneys at the midline.
See Troubleshooting.
-
-
20. Transfer the kidneys onto the transwell filter in the prepared culture dish (from Step 6) using a 200-μL micropipettor with a wide-bore tip or a standard tip with the end cut off.
Excess PBS may be removed with a pipette or allowed to evaporate after the dish is placed in an incubator. The kidney should be located at the air-liquid interface and should not be covered by medium. Allowing space for growth, up to five kidneys can be placed on each filter (Fig. 1D).
-
21. Place the culture dish in a fully humidified 37°C incubator with 5% CO2.
-
22. Culture the kidneys for up to 7 d. Change the medium every 24-48 h, and monitor kidney growth under a dissecting/live imaging microscope daily.
When using a mouse line that expresses a fluorescent marker, kidneys should be exposed to a minimum amount of light while imaging for best results.
To illustrate nephron formation, we have cultured kidneys from Hes1-GFP transgenic mice (Ohtsuka et al. 2006) and taken live brightfield and fluorescent images at time (t) = 0 (Fig. 3K,L) and t = 2 d (Fig. 3M-O). The first GFP+ immature nephrons are evident at E12.5 (Fig. 3L). Distinct developmental structures such as the branching ureteric bud, metanephric mesenchyme, renal vesicles, and S-shaped body are readily distinguished using brightfield imaging (Fig. 3M,N). Hes1-GFP at 2 d of culture demonstrates the emergence of many nephrons (Fig. 3O).
See Troubleshooting.
Whole-Mount Immunohistochemistry of Cultured Kidneys
-
23. To fix the kidneys, transfer the filter into a six-well dish containing cold, freshly prepared 4% paraformaldehyde in PBS. Incubate with gentle rocking for 1 h at 4°C.
The filter will float on top of the fixative and must be submerged manually using forceps. This should be done carefully because the kidneys may detach from the filter at this point.
-
24. Wash the kidneys with gentle rocking in PBST three times for 5 min each at room temperature.
-
25. Transfer the filter to a 24-well dish. Incubate with 200-250 μL of PBS-BB (blocking solution) for at least 1 h at room temperature.
-
26. Remove the PBS-BB with a vacuum aspirator. Add 200-250 μL of primary antibody diluted in PBS-BB. Incubate overnight at 4°C.
-
27. Transfer the filter to a six-well dish and wash extensively with PBST: that is, wash with rocking three times for at least 1 h each at room temperature.
We generally extend the final wash overnight at 4°C.
-
28. Transfer the filter to a 24-well dish. Incubate with 200-250 μL of secondary antibody diluted in PBS-BB for 1 h at room temperature.
From this step forward, signal intensity may be improved by keeping the filters covered or in the dark.
-
29. Transfer the filter to a six-well dish. Wash with rocking in PBST three times for 30 min each at room temperature.
-
30. Mount the kidneys on slides:
-
i. Transfer the filter to a slide (up to two filters per slide).
-
ii. Remove excess PBST from the slide.
-
iii. Place a small drop of Vectashield adjacent to the filter.
-
iv. Cover the filter with a coverslip, allowing the mounting medium to spread as the coverslip lies flat.
Care should be taken to avoid bubbles in the medium because they will interfere with microscopy. Kidneys will flatten and spread out under the coverslip, but will retain their three-dimensional (3D) architecture.
-
-
31. Perform imaging of whole-mount staining with a conventional fluorescence microscope or confocal microscope (see Fig. 4).
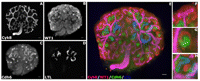 FIGURE 4.
FIGURE 4.Representative four-color immunohistochemistry of cultured kidneys. (A-D) Black and white images of the four markers used in this study. (A) Cytokeratin 8, ureteric bud/collecting duct. (B) WT1, metanephric mesenchyme, podocytes. (C) Cadherin 6, immature and mature proximal tubules. (D) LTL, mature proximal tubules. (E) Four-color merged image of A-D. (F-H) High-magnification images of various stages of nephron formation. WT1-positive nephron stem cells surround the branched ureteric bud tips (F). These stem cells undergo mesenchyme-to-epithelium transition (MET) to form early nephrons with distinct segments (G). Continued growth and differentiation result in segmented tubular epithelium patterned along a proximo-distal axis (H). Scale bar = 100 microns.
Owing to the thickness of the culture, an instrument capable of optical sectioning (i.e., a confocal microscope or microscope equipped with an ApoTome) is recommended.
See Troubleshooting.
TROUBLESHOOTING
Problem: The mouse is not pregnant.
[Step 7]
Solution: In most strains of mice, pregnancy is visible by E11.5 or E12.5. Look for characteristic bulging just above the hindlimb (Fig. 2A). This is especially true for outbred strains (such as CD1), which breed well and have large litters.
Problem: The small size of the embryos/organs makes successful isolation difficult.
[Steps 17-19]
Solution: The dissection technique detailed here will likely require practice to master. If initial attempts yield unsatisfactory results, two possible practice techniques are suggested:
-
1. The same basic technique can be used to isolate kidneys up to E16.5 in the mouse. Practice with older animals and work backward to become comfortable with the technique.
-
2. Rats have essentially the same anatomy as mice at this stage, except they are larger. Timed pregnant rats can be obtained through Harlan Inc. and used for practice. Keep in mind that the gestational age of rats is +3 compared to mice, so an E15.5 rat is equivalent to an E12.5 mouse.
Problem: Kidney grows through the transwell filter.
[Step 22]
Solution: Ensure that the pore size on the filters you are using is correct. Generally, pore size should not be above 1.0 μm.
Problem: Growth and/or survival of kidneys are unsatisfactory.
[Step 22]
Solution: Although we do not routinely encounter problems with growth in serum-free medium, it is possible to supplement the medium with 5%-10% fetal bovine serum (FBS) to improve performance of kidneys in culture.
Problem: The cultures are contaminated.
[Step 22]
Solution: Consider the following:
-
1. Ensure that all solutions and tools are sterile by autoclaving or sterile filtration.
-
2. Carry out dissection on a dissecting microscope placed in a laminar flow hood.
-
3. We do not recommend the addition of antibiotics to the medium. The primary contaminant of cultures is yeast, which will not be eliminated by standard penicillin/streptomycin inclusion.
Problem: There is excessive background staining.
[Step 31]
Solution: Consider the following:
-
1. As with all immunostaining techniques, optimal dilutions for each primary/secondary combination should be determined empirically. Reducing antibody concentrations (especially primary) to the lowest possible level should help to reduce background.
-
2. The inclusion of 10% serum (in PBS-BB) from the species in which the secondary antibody was raised during the blocking step also decreases nonspecific binding of the fluorescent antibody.
-
3. We routinely wash the cultures for longer than the minimum times indicated in the method. Both the primary and secondary washes can be extended overnight.
DISCUSSION
This protocol describes how to isolate, culture, and fluorescently immunostain mouse E12.5 embryonic kidneys. The growth and development of the kidneys in the ex vivo organ culture recapitulates early in vivo development, and critical events occurring at these stages can be studied successfully using this method. Daily monitoring of culture growth should demonstrate a progressive increase in kidney size. This is evident in the continued branching of the ureteric bud and emergence of tubule structures that appear as clusters in between the branching bud (Fig. 3M,N). In the example presented here, an increase in Hes1-GFP, a transgenic marker of newly forming nephrons, was apparent (cf. Fig. 3L to Fig. 3O). The whole-mount immunostaining protocol allows detection of early markers and genes of interest in an informative 3D setting. In our experience, it is amenable to a wide variety of primary antibodies. Here, we demonstrate robust staining of the ureteric bud and developing collecting system (Cytokeratin 8) (Fig. 4A), metanephric mesenchyme and podocytes (WT1) (Fig. 4B), early renal vesicle and proximal tubule (Cadherin 6) (Fig. 4C), and mature proximal tubules (LTL) (Fig. 4D). High-magnification images nicely delineate WT1-positive metanephric mesenchyme surrounding a branched ureteric bud tip (Fig. 4F), newly forming nephrons with distinct segments (Fig. 4G), and mature nephrons with tubular characteristics (Fig. 4H). In addition to whole-mount staining, kidneys can also be embedded in paraffin or OCT medium using standard techniques and sectioned.
The ability to monitor development in culture allows investigators not only to study a wide range of developmental mechanisms, such as branching morphogenesis and early nephron differentiation, but also to image these events in real time. Furthermore, in genetic models that result in embryonic lethality shortly after the onset of kidney development (as often occurs with mutations affecting heart formation), this culture system allows nephrogenesis to be studied independently of confounding phenotypes. It is important to note that this system has limitations. For instance, none of the later events that occur during kidney development, such as cortico-medullary patterning, complete vascularization of glomeruli, or renal filtration, occur in the cultured kidney. Nonetheless, ex vivo kidney culture remains a powerful tool to dissect early events during kidney development. In combination with new methods for the direct manipulation of individual genes or signaling pathways in cultured kidneys, the system described here will allow rapid analysis of a large number of genes (such as candidates generated from gene profiling) without the cost and time required to generate transgenic or knockout animals.
ACKNOWLEDGMENTS
We thank Dr. Raphael Kopan for scientific insight and critical evaluation of the manuscript. The Cadherin 6 antibody was a generous gift from Dr. Greg Dressler. H.B. and S.C.B. are supported by National Institutes of Diabetes and Digestive and Kidney Diseases (NIDDK) grant DK066408. S.C.B. was also supported by NIDDK grant 5T32DK007126. The fluorescent dissecting microscope and live imaging equipment are part of the organogenesis core of the O’Brien Center at Washington University, supported by NIDDK grant 5P30DK079333. The Zeiss Axio Imager.Z1 microscope and ApoTome are part of the Developmental Biology Histology Core at Washington University.








