
NanoCAGE: A High-Resolution Technique to Discover and Interrogate Cell Transcriptomes
- ↵1Corresponding author (rgscerg{at}gsc.riken.jp).
INTRODUCTION
Cap analysis gene expression (CAGE) is a method to identify the 5′ ends of transcripts, allowing the discovery of new promoters and the quantification of gene activity. Combining promoter location and their expression levels, CAGE data are essential for annotation-agnostic studies of regulatory gene networks. However, CAGE requires large amounts of input RNA, which usually are not obtainable from highly refined samples such as tissue microdissections or subcellular fractions. The nanoCAGE method can capture the 5′ ends of transcripts from as little as 10 ng of total RNA and takes advantage of the capacity of current sequencers to produce longer (50-100 bp) reads. The method prepares cap-selected cDNAs ready for direct sequencing of their 5′ ends (optionally mate-paired with the 3′ end) that can provide information about downstream sequences. This protocol describes how to prepare nanoCAGE libraries from as little as 50 ng of total RNA within two working days. The libraries can be sequenced using an Illumina Gene AmplifierIIx with a level of sensitivity 1000 times higher than CAGE.
RELATED INFORMATION
Instead of the cap-trapper method (Carninci et al. 1996, 1997), the nanoCAGE protocol uses a template-switching method based on the reverse transcription of the cap of the messenger RNA (mRNA) to enrich for 5′ ends (Chenchik et al. 1998), as well as a semisuppressive polymerase chain reaction (PCR) approach (Plessy et al. 2010) to minimize short PCR artifacts (Fig. 1). The protocol for nanoCAGE library preparation described here has been comprehensively revised (Table 1) and simplified from the original (Plessy et al. 2010) and no longer uses the time-consuming enzymatic cleavage step that limited the informative length of the tags to ~25 bp.
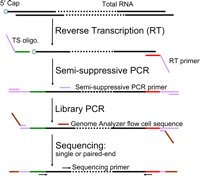
Flowchart of nanoCAGE protocol. Template-switching oligonucleotide (TS) and reverse transcription (RT) primer are added to the first-strand cDNA synthesis reaction. The three guanosine ribonucleosides at the 3′ end of the template-switching primer hybridize in a cap-dependent manner to cytidine deoxynucleosides added to the 3′ end of the newly synthesized cDNA strand by the reverse transcriptase (Hirzmann et al. 1993). After hybridization, the reverse transcriptase extends the cDNA strand using the template-switching oligonucleotide as a template. Hence, the cDNA originating from a capped RNA will have a 3′ end sequence reverse-complementary to the sequence of the template-switching oligonucleotide. These sequences are necessary to synthesize and amplify the second cDNA strand by semisuppressive PCR, which minimizes amplification of shorter artifacts (e.g., primer dimers or aberrant cDNAs template-switched from an RT primer or reverse-transcribed from a TS oligo). End sequences required for sequencing using a Genome AnalyzerIIx are introduced by “library PCR,” and the cDNAs are sequenced as single or paired ends.
Amounts of total RNA ranging from 10 to 1250 ng can be used for nanoCAGE library preparation. For best results, use at least 50 ng of total RNA; this protocol is optimized for that amount. For samples where ~50 μg of RNA is available, the original CAGE method (Kodzius et al. 2006) might be preferable, because it is free from PCR bias. For samples where a few hundreds of nanograms of RNA are available, the preparation of technical replicates is recommended for use as a backup, especially if the analysis is centered on the use of the expression levels.
Libraries prepared using different reverse transcriptases require different conditions for efficient template switching; the reverse transcription of the cap on which the switching depends could play a significant role in these differences (Plessy et al. 2010). Comparisons of the fraction of aligned reads corresponding to the ribosomal RNA (rRNA; <10%) and of redundancy (less than three; see Table 1) showed that libraries prepared with PrimeScript contain approximately twice as many reads aligned to promoter regions (as determined from RefSeq gene models) as those prepared using SuperScript III (Fig. 2).
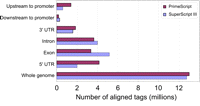
Selection of reverse transcriptase for nanoCAGE library preparation. SuperScript III (SSIII) and PrimeScript (PS) give a higher yield of nanoCAGE cDNAs (data not shown). Libraries were evaluated based on the percentage of aligned reads corresponding to the rRNA and their redundancy (see Table 1 for details). SSIII worked best at 50°C in the presence of sorbitol and trehalose, whereas PS worked best at 40°C in the presence of betaine. Using conditions optimized for each enzyme, SSIII and PS were compared further after alignment of the reads to RefSeq gene models.
CAGE libraries can be used for different types of analyses (Carninci 2010), e.g., promoter discovery (particularly when the available transcript annotation for a tissue or a species is rudimentary), differential expression analysis (Valen et al. 2009), inference of transcription-factor binding sites (Vitezic et al. 2010), or gene networks (Suzuki et al. 2009). Libraries prepared with the nanoCAGE protocol can be paired-end sequenced as CAGEscan libraries (Plessy et al. 2010) that link transcript 5′ ends to downstream regions assembled from the mated 3′ ends.
MATERIALS
Reagents
Agarose gel (1%)
Agencourt AMPure XP (Beckman Coulter A63881)
Agencourt RNAClean XP (Beckman Coulter A63987)
Betaine (5 M; e.g., Wako 023-10862)
DNA loading dye (6X; e.g., MassRuler; Fermentas R0621)
DNA marker: FastRuler DNA ladder, middle range (Fermentas SM1113)
dNTP mixture (2.5 mM; TaKaRa Bio 4030)
Ethanol (70%)
Ex Taq Hot Start Version (TaKaRa Bio RR006A)
High Sensitivity DNA Reagent Kit (Agilent Technologies 5067-4626)
Oligonucleotide, DNA/RNA template-switching, desalted: 5′-TAGTCGAACTGAAGGTCTCCAGCA(rG)(rG)(rG)-3′ (1 mM; Integrated DNA Technologies)
All of the bases are deoxynucleotides except for the guanosine ribonucleotides (rG). These are essential and cannot be replaced by deoxyriboguanosines. Although tests with pure deoxyribo-oligonucleotides for template switching could amplify some libraries, the tags obtained did not identify promoters. To multiplex libraries in single sequencing lanes, a sequence identifier (“barcode”) can be inserted 5′ of the riboguanosines.
Oligonucleotide, library PCR, forward, desalted (Invitrogen): 5′-AATGATACGGCGACCACCGAGATCTACACTAGTCGAACTGAAGG-3′
Oligonucleotide, library PCR, reverse, desalted (Invitrogen): 5′-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT-3′
Oligonucleotide, reverse transcription primer, desalted: 5′-TAGTCGAACTGAAGGTCTCCGAACCGCTCTTCCGATCTNNNNNN-3′ (100 μM; Invitrogen)
The use of oligo-dT reverse transcription primers is not recommended, because they will preferentially amplify short transcripts that originate from 3′ promoters (see Fig. 4B,C in Carninci et al. 2006).
Oligonucleotide, forward second-strand PCR, desalted (Invitrogen): 5′-TAGTCGAACTGAAGGTCTCCAGC-3′
Oligonucleotide, reverse second-strand PCR, desalted (Invitrogen): 5′-TGACGTCGTCTAGTCGAACTGAAGGTCTCCGAACC-3′
Oligonucleotide, sequencing primer, forward, desalted (Invitrogen): 5′-TAGTCGAACTGAAGGTCTCCAGCA-3′
Oligonucleotide, sequencing primer, reverse, desalted (Invitrogen): 5′-CGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT-3′
Reverse transcriptase (200 U/μL; e.g., PrimeScript; TaKaRa Bio 2680A)
The use of PrimeScript for reverse transcription is strongly recommended: Tests of this protocol using different commercially available reverse transcriptases found that PrimeScript was the best for nanoCAGE library preparation (Fig. 2).
RNA samples, total (50 ng/μL)
Appropriate methods should be used to extract (e.g., Trizol LS) and purify (e.g., PureLink RNA Micro Scale Kit [Invitrogen 12183016]) total RNA for nanoCAGE library preparation. Nuclease-free water (or a suitable elution buffer provided with the RNA extraction kit) should be used to elute the RNA. Test-purified total RNA (>50 ng) using an Agilent Bioanalyzer or a NanoDrop spectrophotometer. We recommend total RNA samples with an RNA integrity number (RIN) value ≥7. Store total RNA samples at -80°C until used.
RNaseZap (Ambion 9780)
Sorbitol/trehalose stock solution
SYBR Premix Ex Taq (TaKaRa Bio RR041A)
The product includes the SYBR Premix Ex Taq (Perfect Real Time) (2X) and the ROX Reference Dye II (50X).
Tween 20 (Sigma P9416) (0.1% [v/v], prepared in nuclease-free water)
Water, nuclease-free (e.g., Invitrogen 10977-015)
Equipment
Bioanalyzer (e.g., Agilent 2100; Agilent Technologies)
Centrifugal concentrator (e.g., TOMY)
Centrifuge, benchtop, adapted for use with 96-well plates (e.g., Allegra X-12R; Beckman Coulter)
Electrophoresis unit (e.g., Mupid-α or equivalent)
Gel maker set
Genome AnalyzerIIx (Illumina)
This protocol was designed for use with an Illumina platform. However, it can be adapted for other sequencing platforms by designing suitable primers.
Gloves
Ice-water bath
Imaging system (e.g., BioDoc-It; UVP)
Magnetic stand for bead separation (e.g., Dynal)
Micropipettors
PCR plates, 96-well (e.g., μltraAmp; Sorenson BioScience)
PCR seals, adhesive (e.g., 4ti-0500; 4titude)
PCR system, real-time (e.g., StepOnePlus; Applied Biosystems)
Pipette tips (e.g., Mμltifit; Sorenson BioScience)
Sealant applicator (e.g., PA1; 3M)
Spectrophotometer (e.g., NanoDrop 1000 [Thermo Fisher] or equivalent)
Thermocycler
Tubes, microcentrifuge, low-retention, 1.5-mL and 0.6-mL
Tubes, PCR, siliconized, 0.2-mL (e.g., FastGene)
Water purification system (e.g., Milli-Q Advantage A10; Millipore)
METHOD
Before investigating valuable samples, beginning investigators should practice making control libraries with some available total RNA to develop their abilities to check cDNA synthesis by real-time PCR, to visualize semisuppressive PCR smears on gels, and to interpret Bioanalyzer profiles of prepared libraries.
First-Strand cDNA Synthesis
This process produces first-strand cDNAs from total RNA by random priming; the 5′ ends are captured through template switching. Perform all procedures in RNase-free conditions on a workbench dedicated only to RNA work.
-
1. Mix 8 μL of sorbitol/trehalose stock solution, 1 μL of 100 μM reverse transcription primer, and 1 μL of 1 mM template-switching DNA/RNA oligonucleotide.
Sorbitol/trehalose solutions are viscous; mix very carefully by pipetting at least 10 times.
-
2. Aliquot 1 μL each of the primer solution from Step 1 into separate 0.2-mL siliconized PCR tubes for each sample and for a negative control.
-
3. Add 1 μL of total RNA (i.e., 50 ng) into the sample tubes. Add 1 μL of nuclease-free water into the control tube. Mix the solutions several times for complete homogenization.
If the total RNA is in a larger volume, reduce to 2 μL by centrifugal evaporation at room temperature (to avoid RNA degradation). The presence of sorbitol/trehalose will slow evaporation as the concentration increases, reducing the risk of accidentally drying the RNA.
-
4. Incubate the solutions at 65°C for 10 min in a thermocycler.
-
5. Prepare 8 μL of the reverse transcription reaction mixture per reaction:
-
2 μL of 5X PrimeScript buffer
-
2.5 μL of 2.5 mM dNTPs
-
1 μL of 0.1 M DTT
-
1.5 μL of 5 M betaine
-
1 μL of 200 U/μL PrimeScript
Reverse transcription reactions in the presence of betaine produce better nanoCAGE library outputs (Table 1).
-
-
6. Pause the PCR machine at 22°C. Snap-cool the samples (from Step 4) in an ice-water bath for 2 min.
RNAs must not refold, and cooling occurs faster in an ice-water bath than on ice.
-
7. Return the samples to 22°C in the thermocycler.
-
8. Add 8 μL of the reverse transcription reaction mixture (from Step 5) to the samples.
-
9. Incubate for 10 min at 22°C, 30 min at 40°C, and 15 min at 75°C in the thermocycler.
-
10. Immediately after finishing the reaction, snap-cool in an ice-water bath for 2 min.
Checking the reverse transcription is not recommended at this step, because this will reduce the amount of cDNAs for PCR amplification and therefore increase the number of cycles needed.
-
11. Purify the first-strand cDNAs:
This removes smaller artifacts and primer dimers from the cDNAs that can make it difficult to determine the number of PCR cycles required for semisuppressive PCR. Again, checking the purification at this step is not recommended.
-
i. Use an Agencourt RNAClean XP kit according to the manufacturer’s instructions; mix the cDNA and beads with pipettes.
-
ii. Wash with 100 μL of 70% ethanol.
Before eluting, remove as much ethanol as possible. Do not let the beads dry, because this can reduce recovery.
-
iii. Elute with 40 μL of nuclease-free water.
-
Quantitative Real-Time PCR
Quantitative real-time PCR is performed on a small scale to determine the number of cycles required for large-scale synthesis. This is necessary to keep the number of PCR cycles as low as possible (to reduce bias) and to suppress the synthesis of smaller artifacts.
-
12. For each sample, prepare 8.5 μL of real-time PCR mixture:
-
5 μL of 2X SYBR Premix Ex Taq
-
0.1 μL of 10 μM forward second-strand PCR primer
-
0.1 μL of 10 μM reverse second-strand PCR primer
-
0.2 μL of 50X ROX Reference Dye II
-
3.1 μL of nuclease-free water
Perform real-time PCR in triplicate for the samples and control.
-
-
13. Prepare one μltraAmp PCR plate. Add 8.5 μL of real-time PCR mixture per well.
-
14. Add 1.5 μL of purified cDNA or negative control (from Step 11.iii) to each well.
-
15. Seal the plate with adhesive PCR seal. Ensure a tight seal with a sealing applicator.
Centrifuge briefly if any solution has splashed above the mixture.
-
16. Perform quantitative real-time PCR using a StepOnePlus system according to the manufacturer’s instructions, with the following conditions:
-
i. 95°C for 1 min
-
ii. 40 cycles of 95°C for 15 sec, 65°C for 10 sec, 68°C for 2 min
-
iii. Hold at 4°C
Use “Comparative Ct (ΔΔCt)” for quantification.
-
-
17. Determine the optimum number of PCR cycles required for second-strand cDNA synthesis:
-
i. Determine the “Cycle threshold: (CT)” values for each sample and control reaction after real-time PCR (Fig. 3).
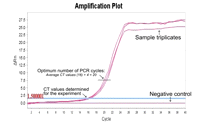 FIGURE 3.
FIGURE 3.Determination of the optimum number of PCR cycles for synthesis of second-strand cDNAs by semisuppressive PCR. A cycle threshold (CT) value is determined by quantitative real-time PCR as the number of cycles required for the fluorescent signal to cross the threshold, i.e., to exceed background levels. The optimum number of cycles for large-scale synthesis is the average CT value plus four.
See Troubleshooting.
-
ii. Calculate the average CT values for each sample and for the negative control.
The average CT value for purified cDNA usually is ~16-20, depending on the source of RNA.
-
iii. Add 4 to the average CT value.
The optimum cycle number should be lower than 25 for the samples and very low or no signal for the negative control.
-
Second-Strand cDNA Synthesis and Amplification by Semisuppressive PCR
This procedure synthesizes the nanoCAGE cDNA libraries.
-
18. Prepare 85 μL of semisuppressive PCR mixture per reaction:
-
10 μL of 10X Premix Ex Taq Hot Start buffer
-
8 μL of dNTPs (2.5 mM each)
-
1 μL of 10 μM forward second-strand PCR primer
-
1 μL of 10 μM reverse second-strand PCR primer
-
0.5 μL of 5 U/μL Premix Ex Taq Hot Start
-
64.5 μL of nuclease-free water
Amplify purified samples in duplicate; a single 100-μL sample is sufficient for the purified control.
The competition between primer annealing and self-annealing is the key mechanism of PCR suppression (Chenchik et al. 1998). The concentration of the primers is thus lower than usual to strengthen the suppressive effect.
-
-
19. Add 85 μL of semisuppressive PCR mixture to each PCR tube.
-
20. Add 15 μL of purified cDNA or negative control (from Step 11.iii) to each PCR tube.
Store the remaining purified cDNAs at -20°C.
-
21. Perform semisuppressive PCR using the following conditions:
-
i. 95°C for 1 min
-
ii. n (optimum PCR cycle number) cycles of 95°C for 15 sec, 65°C for 10 sec, 68°C for 2 min
-
iii. Hold at 4°C
The optimum cycle number (n, as determined in Step 17.iii) is usually ~20-24 cycles, depending on the source of RNA.
PCR tubes can be left overnight in the PCR machine.
-
Purification of Amplified cDNAs
Perform purification and PCR at different locations and use different equipment to avoid cross-contamination.
-
22. Purify the amplified cDNA libraries:
-
i. For each sample, pool the PCR solutions from the duplicate tubes.
Do not purify the negative control. It will be required for confirmation that the primer dimers and short PCR artifacts were eliminated from the amplified cDNAs (see Step 23.i).
-
ii. Use an Agencourt AMPure XP kit according to the manufacturer’s instructions; mix the cDNA and beads with pipettes.
-
iii. Wash with 600 μL of 70% ethanol.
Before eluting, remove as much ethanol as possible. Do not let the beads dry, because this can reduce recovery.
-
iv. Elute with 30 μL of nuclease-free water.
-
-
23. Confirm PCR amplification and purification by agarose gel electrophoresis:
-
i. Mix 2 μL of the purified nanoCAGE cDNAs with 3 μL of nuclease-free water and 1 μL of 6X loading dye. Mix 5 μL of nonpurified control (see Step 22.i.) with 1 μL of 6X loading dye.
-
ii. Load the mixtures on a 1% agarose gel. Load 4 μL of a DNA marker.
-
iii. Electrophorese until the markers separate in the range of from 100 bp to 2 kb (e.g., for agarose minigels, 15-30 min).
-
iv. Visualize the gel under UV light (see, e.g., Fig. 4).
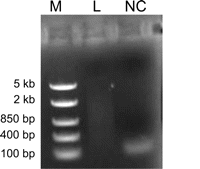 FIGURE 4.
FIGURE 4.Confirmation of semisuppressive PCR amplification and purification. Electrophoresis was performed on a 1% agarose gel at 100 V for 15 min. The purified nanoCAGE library (L) is visible as a smear with small molecular weight artifacts. The nonpurified negative control (NC) is used to visualize the artifacts before removal. (M) DNA ladder, middle range.
Primer dimers or artifacts should not be present in the purified cDNAs. Confirm their elimination by comparing the purified cDNAs with the nonpurified negative control.
See Troubleshooting.
-
-
24. Determine the concentration of the purified cDNAs in triplicate using a NanoDrop spectrophotometer according to the manufacturer’s instructions. Calculate the average value.
The concentration of the purified semisuppressive cDNA libraries should be >20 ng/μL to avoid artificial redundancy (see Discussion).
See Troubleshooting.
-
25. Dilute the samples with nuclease-free water to 10 ng/μL.
Addition of Sequencing Adapters by PCR
This procedure adds adapter sequences to the nanoCAGE libraries for binding to the genome analyzer’s flow cell and for amplification by bridge PCR.
-
26. Prepare 92 μL of PCR mixture per reaction:
-
10 μL of 10X Premix Ex Taq Hot Start buffer
-
8 μL of dNTPs (2.5 mM each)
-
2 μL of 10 μM forward library PCR primer
-
2 μL of 10 μM reverse library PCR primer
-
0.5 μL of 5 U/μL Premix Ex Taq Hot Start
-
69.5 μL of nuclease-free water
Prepare reactions in triplicate for each sample. Also prepare a control reaction without added template.
The concentration of PCR primers is lower than usual because of the small number of cycles, which results in a large leftover.
-
-
27. Add 92 μL PCR reaction mixture to each tube.
-
28. Add 8 μL (i.e., 80 ng) of purified DNA (from Step 25) to each sample tube. Add 8 μL nuclease-free water to the control.
-
29. Perform PCR using the following conditions:
-
i. 95°C for 1 min
-
ii. 1 cycle of 95°C for 15 sec, 55°C for 10 sec, 68°C for 2 min
-
iii. 6 cycles of 95°C for 15 sec, 65°C for 10 sec, 68°C for 2 min
-
iv. Hold at 4°C
-
-
30. Purify the library PCR products:
-
i. For each sample, pool the PCR solutions from the triplicate tubes.
Do not purify the control.
-
ii. Use an Agencourt AMPure XP kit according to the manufacturer’s instructions; mix the cDNA and beads with pipettes.
-
iii. Wash with 900 μL of 70% ethanol.
Before eluting, remove as much ethanol as possible. Do not let the beads dry, because this can reduce recovery.
-
iv. Elute with 30 μL of 0.1% Tween 20 in nuclease-free water.
-
-
31. Determine the concentration of the purified libraries in triplicate using a NanoDrop spectrophotometer according to the manufacturer’s instructions. Calculate the average value.
The concentration of the purified library PCR products is usually >20 ng/μL (typically, ~20-30 ng/μL). Store the prepared nanoCAGE libraries at -20°C until sequenced.
Analysis of nanoCAGE Libraries
It is important to check the removal of PCR primer dimers/artifacts, as well as to know the profile of the amplified libraries (e.g., size distribution, molar concentration) to calculate the amount of molecules to apply in the sequencing reaction.
-
32. Dilute 1 μL of each purified library to 5 ng/μL with 0.1% Tween 20.
-
33. Analyze 1 μL each of the diluted samples using a High Sensitivity DNA Reagent Kit according to the manufacturer’s instructions.
Analyze the libraries in triplicate.
See Troubleshooting.
-
34. Determine the nanomolar concentration of the library from the Bioanalyzer data (Fig. 5):
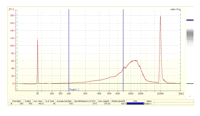 FIGURE 5.
FIGURE 5.Bioanalyzer profile of a typical nanoCAGE library (5 ng/μL). This protocol assumes that only cDNAs in the 200-700-bp range are sequenced efficiently. Longer templates are likely to be outcompeted by shorter ones for binding to the flow cell; they also could be difficult to amplify using the bridge PCR.
This protocol assumes that only cDNAs in the 200-700-bp range are efficiently sequenced in the Gene AnalyzerIIx sequencer. There should be no peak shorter than 150 bp visible on the Bioanalyzer data.
-
i. Open the library DNA analysis file (.xad).
-
ii. Double-click on the electropherogram data of the sample to be analyzed.
-
iii. Click on the “Region table” icon under the page. Scroll into the table.
-
iv. Right-click the mouse button. Select “Modify region.”
-
v. Input 200 and 700 in the boxes “From[bp]” and “To[bp]”, respectively.
-
vi. Click the “OK” button to determine the molarity.
-
vii. Calculate the average concentration of the triplicate assays.
-
viii. Multiply this concentration with the dilution factor (from Step 32) to obtain the original library concentration.
-
-
35. Sequence the nanoCAGE libraries at a final concentration of 15 pM using an Illumina Genome AnalyzerIIx platform for single or paired-end reads.
One lane on this sequencer generally yields more than 15 million reads. Although not tested, a higher number of readings is possible with other systems (e.g., HiSeq2000 [Illumina]).
TROUBLESHOOTING
Problem: CT value is lower than usual (i.e., 16-20).
[Step 17.i]
Solution: Perform a small-scale PCR using the extra purified cDNA solution (from Step 20) as described in Steps 18-21; scale the reaction volumes down to one-fifth (i.e., a final volume of 20 μL). Take aliquots after 20, 22, and 24 cycles. Confirm amplification and detect optimal cycle number by running aliquots of the products on a 1% agarose gel. The number of cycles to be used for the large-scale reaction is defined by the cycle where a signal becomes visible on the gel for a sample, while the negative control still has no detectable products.
Problem: CT value is higher than usual (e.g., >20).
[Step 17.i]
Solution: The most probable causes are partial degradation of RNA, impurities or inhibitory substances in the RNA, reverse transcriptase inactivation, or a high concentration (~50 ng/μL) of the smaller (<200-bp) fraction. Consider the following:
-
1. If possible, recheck the RNA quality with a Bioanalyzer.
-
2. Check the reverse transcriptase using a control template.
-
3. Check for any possible source of contamination during the reverse transcription preparation.
-
4. Perform the reverse transcription reaction within the shortest time possible.
Problem: Primer dimers or short artifacts could not be removed from the library.
[Steps 23.iv and 33]
Solution: This can occur with nanoCAGE libraries begun with <50 ng of total RNA. Repurification using the AMPure XP Kit should solve this problem.
Problem: The concentration of the purified semisuppressive PCR library is <20 ng/μL.
[Step 24]
Solution: Although the library could be sequenced, it might be highly redundant (see Discussion). Therefore, if a backup sample is available, remaking the library is recommended. Alternatively, the library can be used for promoter discovery (see Discussion).
DISCUSSION
The transcripts detected by nanoCAGE are strand specific. Once prepared, the libraries can be sequenced for both 5′-end single and paired-end reads. Single-read sequencing can pinpoint the gene’s product (i.e., the mRNA molecule) from the transcription start site, whereas paired-end sequencing can link start sites to downstream sequences, providing insight into the architecture of transcripts and thus into their possible functions (Plessy et al. 2010). Currently, the ability to fully scan the 3′ end of long transcripts is limited by the length of the paired-end reads, although further developments in sequencing technology could overcome this limitation.
NanoCAGE libraries can be prepared from <50 ng of total RNA, although with a risk of molecular bottlenecks and the need for a higher number of PCR cycles (see below). NanoCAGE libraries have been successfully prepared from nonpolyadenylated or polysomal RNA, RNA extracted from histological sections that were stained by immunohistochemistry, and samples microdissected with laser capture. In general, nanoCAGE will also work on degraded RNA at the progressive expense of its accuracy in detecting promoters. NanoCAGE even detects gene expression using decapped and fragmented samples, albeit similarly to RNA-seq protocols (Plessy et al. 2010). It might require a higher number of PCR cycles (>30) to prepare nanoCAGE libraries from total RNA samples that contain a large fraction of small RNAs (<150 bp), e.g., when their concentration is within an order of magnitude of the oligonucleotides used in the reverse transcription.
Depending on the source of total RNA, the fraction of sequencing reads that align to rRNA can be higher than usual (>8%). Because rRNA is not capped, a high number of reads matching rRNA suggests that the template-switching reaction was not highly specific, and that the detection of the promoters thus will not be accurate at a base-pair level. Some cell lines contain more rRNA than others, and some RNA samples contain a larger fraction of rRNAs (polysomal RNA, nonpolyadenylated RNA, etc.); nanoCAGE libraries prepared from these samples will always contain more reads matching the ribosomal DNA. Conversely, for many organisms, a full-length sequence of the ribosomal DNA-repeated unit is not available, and the percentage of reads matching rRNA will not be an accurate measurement of the efficiency of template switching.
The hallmark of CAGE (Kodzius et al. 2006; Valen et al. 2009; Carninci 2010) is the addition of extra guanosines at the 5′ end of the tags. However, in nanoCAGE these guanosines become part of the 5′ linker, and most of them are removed as linker sequence. Nevertheless, in most cases mismatches at the 5′ end of the tags are expected to be guanosines. Results contrary to this could indicate a problem in the library, e.g., degradation of the starting material resulting in a much lower fraction of capped 5′ ends.
A high redundancy usually indicates a molecular bottleneck, where only a small fraction of the original mRNA molecules contribute reads in the libraries. Libraries where the concentration of purified cDNAs after semisuppressive PCR is <20 ng/μL often have a redundancy higher than 5 (Table 1). This could result from preferential amplification of a subset of templates or by initial rarity of PCR templates, e.g., because of low efficiency in reverse transcription or template switching. Libraries with a high redundancy, nevertheless, can still be used for promoter discovery. Keep in mind, however, that rare transcripts might not appear, and that the shape of the transcription start site could become artificially sharpened, even with broad promoters (Carninci et al. 2006).
The nanoCAGE method has the ability to grasp the complexity of the promoter landscape from fewer cells without the need to rely on predetermined gene models such as RefSeq. NanoCAGE libraries prepared using this protocol can be exploited for new applications in drug screening, biopsy analysis, and whole-transcriptome association studies.
ACKNOWLEDGMENTS
This work was funded by a Grant-in-Aid for Scientific Research (A) 20241047 to P.C., a grant of the 7th Framework of the European Union commission to P.C. (Dopaminet), U.S. National Human Genome Research Institute grant U54 HG004557 to P.C, and Research Grant for RIKEN Omics Science Center from the Japanese Ministry of Education, Culture, Sports, Science and Technology to Y. Hayashizaki. We acknowledge RIKEN GeNAS for sequencing the libraries J53-GA to J55-GA and J61-GA to J65-GA using the Genome AnalyzerIIx, as well as for subsequent data processing.








