
Historical Evolution of Laboratory Strains of Saccharomyces cerevisiae
- Centre for Genetic Architecture of Complex Traits, Department of Genetics, University of Leicester, Leicester LE1 7RH, United Kingdom
Abstract
Budding yeast strains used in the laboratory have had a checkered past. Historically, the choice of strain for any particular experiment depended on the suitability of the strain for the topic of study (e.g., cell cycle vs. meiosis). Many laboratory strains had poor fermentation properties and were not representative of the robust strains used for domestic purposes. Most strains were related to each other, but investigators usually had only vague notions about the extent of their relationships. Isogenicity was difficult to confirm before the advent of molecular genetic techniques. However, their ease of growth and manipulation in laboratory conditions made them “the model” model organism, and they still provided a great deal of fundamental knowledge. Indeed, more than one Nobel Prize has been won using them. Most of these strains continue to be powerful tools, and isogenic derivatives of many of them—including entire collections of deletions, overexpression constructs, and tagged gene products—are now available. Furthermore, many of these strains are now sequenced, providing intimate knowledge of their relationships. Recent collections, new isolates, and the creation of genetically tractable derivatives have expanded the available strains for experiments. But even still, these laboratory strains represent a small fraction of the diversity of yeast. The continued development of new laboratory strains will broaden the potential questions that can be posed. We are now poised to take advantage of this diversity, rather than viewing it as a detriment to controlled experiments.
CONGENIC VERSUS ISOGENIC STRAINS
By far, the most widely used laboratory strain of budding yeast has been S288C (Mortimer and Johnston 1986; Olson et al. 1986; Winston et al. 1995; Brachmann et al. 1998). The history of S288C and its derivatives has been discussed by Mortimer and Johnston (1986); they describe its complicated pedigree, involving the use of various sources of yeast in crosses that were not well-controlled. As we will see below, one of the reasons for its wide use, in addition to the generosity of Mortimer and other yeast researchers (including Fred Sherman, David Botstein, Gerry Fink, Phil Heiter, Francois Lacroute, Fred Winston, and many others), was the fact that isogenic MATa and MATα versions were available through a spontaneous rare MAT switching in the original S288Cα (ho) strain. Two spores from a spontaneous diploid (X2180-1A and X2180-1B) were widely distributed and used, and these allowed the development of isogenic series of strains through crossing (Mortimer and Johnston 1986). Until molecular genetic tools became available, most researchers working with other strains had to deal with varying levels of congenicity through backcrossing to one or another parental input (usually S288C or a congenic derivative). This invariably led to interesting observations as well as problems with controls. One series of congenic strains is YNN216 and derivatives YPH499, YPH500, and YPH501 (Johnston and Davis 1984; Sikorski and Hieter 1989), which have been widely used and are isogenic within the set. Another strain developed in this way was W303, derived from crossing a set of desired markers using S288C and several other strains for several generations (Rodney Rothstein as described at the Saccharomyces Genome Database [SGD]). Although highly related to S288C, 15% of its genetic material from elsewhere, it is phenotypically different from S288C in several respects as will be seen below. Once molecular genetics and the cloning of genes came to exist, the HO endonuclease could be used to create isogenic derivatives of the opposite mating type, as was done for W303, overcoming the issue of variation in congenic derivatives. Genetic combinations of mutants in W303, for example, could be compared with W303 rather than S288C once these isogenic series were available.
The S288C pedigree also gave rise to A364a, another widely used strain, particularly for cell-cycle studies. Although obviously related, these two strains display quite different properties and were used in different types of experiments (Hartwell 1967; Kaiser and Schekman 1990) and use of both has led to Nobel Prizes (2001 and 2013). Table 1 lists some of the popular laboratory strains during the rise of yeast genetics and biology. There are many more described at SGD (see Box 1). In addition to S288C (and derivatives), A364a, and W303, these include FL100, CEN.PK, YNN216, YPH499, YPH500, YPH501, and Sigma1278b. All of these are related in some way to S288C. Other strains such as SK1 and Y55 were derived from independently isolated yeast and have been developed for specific studies, including sporulation for both (Tauro and Halvorson 1966; Kane and Roth 1974; Bishop et al. 1992) and various mutant screens for Y55 (McCusker et al. 1987; McCusker and Haber 1988a,b). More recently, there has been an explosion of independent yeast isolates that have been made genetically tractable for various studies including RM11-1A derived from a vineyard yeast (Brem et al. 2002), YJM789, a clinical isolate (Wei et al. 2007), and the Saccharomyces Genome Resequencing Project (SGRP) clean lineages from Wine/European (WE), North American (NA), West African (WA), Sake (SA), and Malaysian (MA) populations (Liti et al. 2009). Other isolates have been sequenced, such as EC1118 from the wine industry (Novo et al. 2009) and PE-2 from the bioethanol industry (Argueso et al. 2009), but they have yet to be adapted to easy laboratory use. Most recently, there has been an expansion of Saccharomyces cerevisiae diversity with the discovery of new populations from China (Wang et al. 2012). Although not entirely sequenced, the relationships of these new populations to the other known populations of yeast can be seen in Figure 1. Each laboratory strain has an interesting story behind their derivation and use and a few will illustrate important lessons.
BUDDING YEAST RESOURCES
Saccharomyces Genome Database (SGD) contains almost all you need to know about S. cerevisiae, with connections to other data and resources. www.yeastgenome.org
Strain/reagent collections/repositories:
-
American Type Culture Collection (ATCC) maintains yeast stocks and clones. www.lgcstandards-atcc.org
-
EUROSCARF, the EUROpean Saccharomyces Cerevisiae ARchive for Functional analysis, maintains a collection of systematic deletion strains searchable by gene name. web.uni-frankfurt.de/fb15/mikro/euroscarf/
-
National Collection of Yeast Cultures (NCYC) maintains over 3100 nonpathogenic yeasts, including type strains, strains of general interest for education and research, strains of industrial importance, and genetically marked strains. www.ncyc.co.uk
-
Common Access to Biological Resources and Information (CABRI) includes catalogs from European culture collections for yeast and other organisms. www.cabri.org
-
Yeast Genetic Resource Center (YGRC) maintains over 4800 S. pombe strains and over 9000 S. cerevisiae strains. yeast.lab.nig.ac.jp/nig/index_en.html
-
Industrial Yeasts Collection DBVPG is an academic biological resource center that specializes in yeasts and yeast-like microorganisms. www.agr.unipg.it/dbvpg
-
The collections at Centraalbureau voor Schimmelcultures (CBS) offer comprehensive coverage of the culturable biodiversity of the fungal kingdom. www.cbs.knaw.nl
-
Addgene is a nonprofit plasmid repository that distributes many plasmids for yeast research. It includes a collection of Yeast Advanced Gateway Destination Vectors created by Dr Susan Lindquist’s laboratory. www.addgene.org/yeast_gateway
-
Yeast-GFP Clone Collection from Dr Erin O’Shea and Dr Jonathan Weissman at UCSF consists of carboxy-terminal tagged open reading frames (ORFs). clones.lifetechnologies.com/cloneinfo.php?clone=yeastgfp
-
Yeast GST-Tagged Collection for inducible overexpression of yeast ORFs was developed in the Andrews laboratory at the University of Toronto. www.thermoscientificbio.com/non-mammalian-cdna-and-orf/yeast-gst-tagged-orfs
-
Yeast Knockout (YKO) Collection is available from the Saccharomyces Genome Deletion Consortium. www-sequence.stanford.edu/group/yeast_deletion_project/deletions3.html
-
Yeast-TAP Fusion Library from Dr Erin O’Shea and Dr Jonathan Weissman at UCSF contains open reading frames (ORFs) that are tagged with high-affinity epitopes and are expressed from their natural chromosomal locations. www.thermoscientificbio.com/non-mammalian-cdna-and-orf/yeast-tap-tagged-orfs
-
Yeast Tet-promoters Hughes Collection contains 800 essential yeast genes for which expression is regulated by doxycycline. www.thermoscientificbio.com/non-mammalian-cdna-and-orf/yeast-tet-promoters-collection/?redirect=true
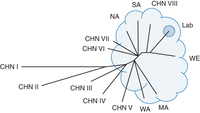
Topology of the phylogenetic relationship among yeast isolates. The relationships of the main lineages of S. cerevisiae isolates are displayed without all strains indicated. The topology shown is adapted from the SGRP analysis of whole-genome sequences (Liti et al. 2009) and the analysis of nine concatenated gene sequences for the Chinese isolates (Wang et al. 2012). The main lineages from the SGRP analysis are North American (NA), Wine/European (WE), Sake (SA), West African (WA), and Malaysian (MA). The Chinese analysis revealed eight new populations (CHN I through VIII), some of which are closely related to the previously known lineages, whereas some are more diverse. The cloud represents the known whole-genome sequence space of S. cerevisiae as a species. The majority of laboratory strains fall into the small circular area within the cloud. The exceptions being SK1 and Y55, which fall close the WA population.
Budding yeast laboratory strains: old and new
CONDITIONAL EFFECTS AND THE SCOURGE OF CONGENICITY
In the 1980s, there were many debates over the requirement of certain genes for certain functions, and one memorable one was whether or not FUS3 (an MAPK kinase) was required for pheromone-mediated signal transduction with an East coast versus West coast of the United States difference in outcome associated with a fus3 mutation. The resolution came down to a difference between W303 and S288C. S288C has a mutation in KSS1, another MAPK kinase with overlapping function, while it is intact in W303 (Elion et al. 1991). The synthetic effect on S288C, in this case, we could say conditional effect, was due to the genetic background difference. Here there is only one relevant difference between the strains. There are many other phenotypic differences between these two strains as well as other related strains, which are due to genetic differences between the backgrounds. At the time of the FUS3/KSS1 story, it was used as a lesson for proper controlled experiments with isogenic strains as well as making sure the genetic context of an experiment was clear. Now we take advantage of such differences to map other genes involved in particular traits. An early example of this was the rediscovery of filamentous growth in yeast, not seen in S288C, due to three genetic differences between S288C and Sigma1278b-related strains (Liu et al. 1993). The difference in the mating pathway between the two strains has recently been elucidated (Chin et al. 2012).
The importance of this conditional effect of mutations dependent on genetic background can be seen best in the comparison of gene knockouts in S288C Sigma1278b, which is closely related to S288C with at least half of the genome identical. When only essential genes are considered, Boone and colleagues (Dowell et al. 2010) found several genes that were essential in one background and not in the other. These conditional essential genes were then found to have more than one modifying genetic difference between the strains underlying the essentiality (Dowell et al. 2010). Individual cases of conditional essential genes were seen in the past such as with the clathrin heavy chain, first reported to be nonessential (Payne and Schekman 1985) and then found to be essential once an unlinked segregating suppressor was found (Lemmon and Jones 1987). Life versus death is the most extreme phenotype, and this observation is only the tip of the iceberg of genetic background effects. Synthetic lethal screen exploits the simplest of these genetic background effects with only two differences to consider and other synthetic interactions between two gene knockouts are the basis of large genetic interactions studies (Tong et al. 2001; Jorgensen et al. 2002; Tong et al. 2004).
EXPANSION OF AVAILABLE STRAINS AND THE EMBRACEMENT OF DIVERSITY
As seen above, the majority of laboratory strains in early use were all related to each other. The exceptions were the independently isolated SK1 (Kane and Roth 1974; Bishop et al. 1992) and Y55 (Tauro and Halvorson 1966; McCusker and Haber 1988a,b). Until recently, it was thought that these isolates, both used in studies of sporulation and meiotic recombination, were very different from each other genetically, as well as from S288C, as they behaved differently phenotypically. We now know through sequencing that these two isolates are indeed related to each other and have segments of their genomes from other populations (Liti et al. 2009). One could speculate that early studies with these strains, Y55 was used for sporulation studies in the Halvorson laboratory (Tauro and Halvorson 1966) and Kane was used in Halvorson’s laboratory before starting his own studies of sporulation using SK1 (Kane and Roth 1974), led to their inadvertent interbreeding.
Through the efforts of many people, there are a great many diverse strains available for use, opening the door to new areas of inquiry. This has been particularly evident in complex trait analysis where the standing genetic variation between strains and their phenotypic effects is embraced as a source of data (Brem et al. 2002; Steinmetz et al. 2002; Sinha et al. 2006; Cubillos et al. 2011; Parts et al. 2011), rather than treated as a source of noise if not worse. As with the above example of the conditional essential genes between two related laboratory strains, most phenotypes are due to complex interactions between more than two genes as well as with the environment.
S288C was the first eukaryotic organism sequenced (Goffeau et al. 1996) enhancing its status as the most widely used yeast strain. This sequence is the reference for many studies including the assessment of genetic variation in other isolates. The genetic diversity and relationships between strains were first gleaned from microarray data (Winzeler et al. 2003; Schacherer et al. 2007; Schacherer et al. 2009). This revealed that there were two possible domesticated populations, for Wine and Sake use (Fay and Benavides 2005), and that the laboratory strains were all related to each other. As sequencing became more efficient, many more strains have been sequenced. At the end of the first-generation sequence era, 36 isolates of S. cerevisiae from a wide variety of sources were sequenced (Liti et al. 2009) revealing that there are at least five “clean” lineages of yeast with laboratory strains and many others being recent mosaic outbreds of two or more of these populations. Second-generation technology has led to the genome sequences of many other strains, and new populations are being discovered that will add to the sequence space of the species (Wang et al. 2012). In Figure 1, we can see the global phylogenetic relationship between the current known groups of S. cerevisiae. Those within the cloud have genetically tractable derivatives (McCusker and Haber 1988a,b; Bishop et al. 1992; Winston et al. 1995; Brachmann et al. 1998; Brem et al. 2002; Cubillos et al. 2009). The new diverse population will likely be amenable to genetic manipulations in the near future. It is clear that the classical laboratory strains represent a small fraction of the available genetic diversity. Future studies may benefit from taking advantage of this increased diversity.
RESOURCES AVAILABLE
This expansion of available strains as well as additional tools to facilitate research has led to a plethora of resources, both in terms of strain collections and in plasmid collections, vectors, and drug markers. In Box 1, the current URLs are provided for a variety of these resources, many of which are also available through the authors of the work or through members of the community using these resources. The font of most knowledge about yeast is of course the Saccharomyces Genome Database, which in addition to containing detailed information about genes, genomes, and phenotypes, has a large section dedicated to community information and resources.
There are a number of culture collections around the world with various yeast strains from diverse origins available (see Box 1 for details). These include the ATCC in the United States, the NCYC in the United Kingdom, the YGRC in Japan, the DBVPG in Italy, and the CBS in The Netherlands. In addition, there is a collection of culture collection catalogs available at CABRI.
There are large collections of yeast strains for systematic studies. Perhaps the most widely used now is the yeast knockout collection, now with more than 20,000 strains available as MATa and MATα haploids, heterozygous knockout diploids (these contain the essential gene deletions, whereas the other sets do not), and homozygous knockout diploids. Created with unique barcodes, flanking the KANMX disruption cassette, these collections are useful for both specific gene studies and global studies. They are all created in one or another of the BY series, isogenic with S288C (Table 1) and have useful auxotrophies for systematic analyses (Shoemaker et al. 1996; Winzeler et al. 1999). They are available from the Saccharomyces Genome Deletion Consortium as well as EUROSCARF and can also be obtained from members of the community. Complementing this collection are over 800 essential genes expressed under a regulatable promoter (also in the BY strain background) (Mnaimneh et al. 2004; Davierwala et al. 2005) (Box 1), which facilitates the study of loss of function of these essential genes. There are also ways to generate temperature-sensitive alleles of essential genes for functional studies (Ben-Aroya et al. 2010; Li et al. 2011).
Complementing the deletion collections are various sets of tagged proteins for a variety of studies. Over 4150 proteins have been tagged with GFP in BY4741 for localization studies (Box 1) (Huh et al. 2003). A similar collection of TAP fusions of over 4250 proteins in BY4741 is also available for high-affinity purification and protein complex studies (Ghaemmaghami et al. 2003). Over 5000 of the genes have been GST-tagged for overexpression studies as well, also in BY4741 (Sopko et al. 2006).
Other resources include the collection of each gene under their native promoter on CEN plasmids, each with a molecular barcode (the MoBY collection) (Ho et al. 2009). This is useful for both individual gene studies and systematic complementation studies to identify mutations. Functional studies that go beyond use of the collections already described are facilitated by various vector collections including the 288 yeast advanced gateway vectors (Box 1) (Alberti et al. 2007).
WHAT STRAIN(S) TO USE
In the past, the choice of strain depended in large part on the markers available (Barnett 2007). Early genetic markers were auxotrophies generated through spontaneous or induced mutagenesis. The advent of molecular techniques allowed the creation of specific targeted mutations and deletions, and in recent years, the use of dominant drug-resistant markers adapted for use in yeast has made virtually any strain amenable to genetic manipulation by creation of appropriate gene knockouts (HO, for example, to create stable haploids, or specific auxotrophies such as ura3). Auxotrophies that continue to be used for numerous experiments are ura3, lys2, trp1, his3, and leu2, but there are many others used for various purposes. The major drug marker used confers G418 resistance and is generally taken from the KANMX cassette created for gene knockouts in yeast (Wach et al. 1994). There are several other marker cassettes available as well (Goldstein and McCusker 1999; Goldstein et al. 1999). The problem of limited marker availability, when multiple knockouts are required, has been overcome with various recycling schemes. A recent advance is the use of modified loxP sites and dominant drug markers such that any strain can be genetically modified (Carter and Delneri 2010), which opens the door to the diverse wild populations that may harbor interesting genetic and phenotypic variation.
The choice of strain also depends on the questions being addressed (Barnett 2007). The ease of genetic and molecular manipulation today makes it feasible to do experiments, even high-throughput global experiments, in virtually any and perhaps in multiple strain backgrounds. We now know that rather than being a problem, differences in outcome between different genetic backgrounds can be highly informative. For many questions, the workhorse of yeast genetics, S288C, and all the collections derived from it, may be the best solution. It is usually the most efficient solution as all the hard work and infrastructure are already in place. However, S288C is not the best representative of the species and other isolates may be worth the effort to work up for some experiments. For example, there are at least six well-studied gene families not present in S288C and 38 new gene families not present in S288C were found in a population genomics survey (Liti et al. 2009).
For many studies, the deletion collection and resources available for S288C are probably the first choice as there is a tremendous amount of information and experimental data available for help in particular studies as well as for comparison. However, this workhorse is not always the best for all studies and in particular studies of variation such as quantitative genetics of complex traits or in functional analysis of genes not present in its genome. Some studies would be better using more industrially relevant strains such as the commonly used Ethanol Red (available in many places and free to use without restrictions) or the wine strain EC1118 (Novo et al. 2009) which has much better fermentation properties than S288C. Perhaps the most appropriate approach to strain choice is to screen a variety of strains first for the particular study of interest. Choose ones that behave best under the conditions of the experiment before investing a great deal of time. Simple things like growth rates under various conditions, flocculation, temperature sensitivity (cold and hot), mating ability, etc., can be done easily before undertaking an experiment. As it is the differences between individuals that makes biology interesting, it may be wise to choose several strains in a comparative study as these differences should be embraced (Liti and Schacherer 2011; Nieduszynski and Liti 2011).
FUTURE PROSPECTS
Saccharomyces cerevisiae remains at the forefront of biology as a model organism. This is in large part because of the tools and resources that facilitate and enable sophisticated experiments. As functional studies go beyond the S288C laboratory strain, new tools and collections will become useful. Entire deletion collections in other strain backgrounds will become useful, facilitating the study of conditional phenotypes (Dowell et al. 2010) as well as in reciprocal hemizygosity confirmation of candidate QTLs (Cubillos et al. 2011). There is even an argument for deletion collections in the closely related species. Other tools such as tagged proteins and ORF collections on plasmids in other strain backgrounds will also be useful to the community. Finally, it is clear that a great deal of phenotypic variation comes from presence/absence of genes and gene families as well as copy-number variation (Bergstrom et al. 2014), and this genomic variation, mostly in the subtelomeres, is underexplored. As sequencing becomes less expensive (Wilkening et al. 2013) and new strains are identified (Wang et al. 2012), the choice of strains to use will become both more informed and more difficult due to the huge number of possibilities. In contrast, the prospects of the synthetic yeast genome may remove the need for many of these tools and resources as specific strains for specific studies could be synthesized as needed (Annaluru et al. 2014).
- © 2016 Cold Spring Harbor Laboratory Press








