
High-Throughput Quantitative Genetic Interaction Mapping in the Fission Yeast Schizosaccharomyces pombe
- 1Department of Cellular and Molecular Pharmacology, University of California San Francisco, San Francisco, California 94518
- 2Systems Biology Ireland, University College Dublin, Belfield, Dublin 4, Ireland
- 3North West Cancer Research Institute, Bangor University, Bangor LL57 2UW, United Kingdom
- ↵4Correspondence: roguev{at}gmail.com; nevan.krogan{at}ucsf.edu
Abstract
Epistasis mapping, in which the phenotype that emerges from combining pairs of mutations is measured quantitatively, is a powerful tool for unbiased study of gene function. When performed at a large scale, this approach has been used to assign function to previously uncharacterized genes, define functional modules and pathways, and study their cross talk. These experiments rely heavily on methods for rapid sampling of binary combinations of mutant alleles by systematic generation of a series of double mutants. Epistasis mapping technologies now exist in various model systems. Here we provide an overview of different epistasis mapping technologies, including the pombe epistasis mapper (PEM) system designed for the collection of quantitative genetic interaction data in fission yeast Schizosaccharomyces pombe. Comprising a series of high-throughput selection steps for generation and characterization of double mutants, the PEM system has provided insight into a wide range of biological processes as well as facilitated evolutionary analysis of genetic interactomes across different species.
EPISTASIS MAPPING
Genetic epistasis is classically defined as a biological phenomenon whereby the phenotype associated with a perturbation (point mutation or deletion) of a gene is modulated by a perturbation in another gene. Such relationships between genes are broadly termed genetic (or epistatic) interactions (GIs) and have long been used as a tool to dissect the functional relationships among sets of genes (Kaiser and Schekman 1990; Guarente 1993). Growth rate as a proxy for fitness is commonly used to assess GIs manifesting themselves as strong qualitative differences in growth rates between the observed and expected phenotype of the double mutant given the phenotypes of the two single mutants. Based on this, two distinct flavors of GIs can be defined. A type of interaction referred to as synthetic sickness/lethality is observed when two mutations do not cause severe growth defects by themselves but, when combined, result in severely compromised fitness or death. Synthetic sickness/lethality, or negative genetic interactions, point to two genes acting in independent but complementary parallel pathways (Hartman et al. 2001; Costanzo et al. 2010). Conversely, a situation in which a normally deleterious mutation loses its impact in the context of a second mutation is referred to as phenotype suppression or positive genetic interaction. Positive GIs often identify genes that act in the same pathway or as part of the same protein complex or functional module (Collins et al. 2007; Roguev et al. 2008; Ryan et al. 2012). These two classes of GIs have been extremely useful in deconvoluting the topology and organization of molecular pathways in model organisms (Fig. 1A; Collins et al. 2007; Roguev et al. 2008; Wilmes et al. 2008; Fiedler et al. 2009; Aguilar et al. 2010; Costanzo et al. 2010). Unlike protein–protein interactions, which are limited to physically interacting gene products, GIs report on functional relationships and reveal how groups of genes and functional modules act in concert to carry out higher-level biological functions. Additionally, GIs describe the cross talk between pathways and processes (Beltrao et al. 2010). Epistasis maps are therefore a natural complement to protein–protein interaction maps, and integration of these two types of information has proven to be extremely powerful in understanding complex biological phenomena in various model systems (Kelley and Ideker 2005; Keogh et al. 2005, 2006; Collins et al. 2007; Bandyopadhyay et al. 2008; Roguev et al. 2008; Fiedler et al. 2009; Hannum et al. 2009; Beltrao et al. 2010; Frost et al. 2012; Ryan et al. 2012).
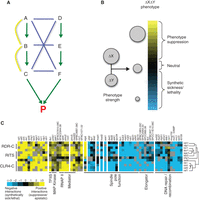
Epistasis mapping overview. (A) The logic behind synthetic sick/lethal (negative, blue edges) and buffering (positive, yellow edges) interactions. Two parallel pathways (A–B–C and D–E–F) lead to the same essential product (P). Different types of genetic interactions emerge, depending on how genes are disrupted in pairs. (B) Modeling genetic interactions, capturing the continuous spectrum of phenotype strengths. Single deletions (Δ of genes X and Y) are combined in a double-deletion strain. The expected fitness defect of the double mutant (ΔXΔY) is the product of the fitness defects of ΔX and ΔY. Genetic interactions are modeled as deviations from this expected value. The size of the circle is proportional to fitness. (C) Computational analysis of epistatic interactions. GI profiles of 12 members of the RNAi machinery in S. pombe are clustered. Positive interactions are observed within the pathway and with parts of the transcription machinery. Negative interactions are observed with genes involved in spindle function, DNA repair, and the elongator complex.
Technologies that allow generation of large numbers of double mutants in a systematic and parallel fashion are now available in bacteria (Butland et al. 2008; Typas et al. 2008), budding yeast (Tong et al. 2001; Pan et al. 2004), fission yeast (Roguev et al. 2007; Dixon et al. 2008), Caenorhabditis elegans (Lehner et al. 2006; Byrne et al. 2007), Drosophila melanogaster (Horn et al. 2011), and mammalian cells (Bassik et al. 2013; Roguev et al. 2013). In yeast, high-throughput GI mapping was pioneered in Saccharomyces cerevisiae with the development of two key experimental platforms—synthetic genetic array (SGA) (Tong et al. 2001) and diploid-based synthetic lethality analysis on microarrays (dSLAM) (Pan et al. 2004). Both use a library of deletion mutants and a specially engineered genetic background, allowing for selection of double mutants in bulk. An SGA experiment is usually performed using colony arrays on agar plates. Using array replicating robotics, a query gene deletion is crossed systematically with a whole-genome array of deletion mutants. The diploid arrays are then induced to differentiate through meiosis and sporulation. Through a series of selection steps, arrays of double mutants are then generated and examined for synthetic lethal interactions. dSLAM relies on introducing the query deletion in a barcoded pooled library of diploid heterozygous deletion mutants. After meiosis and sporulation, haploid single and double mutants are selected for and GIs are uncovered through microarray analysis of a growth competition assay.
A distinctive feature of classical SGA is that it employs a binary scoring scheme for phenotype strength (growth vs. no growth) and captures only the extreme cases within a much broader spectrum of interactions. Current state-of-the-art frameworks for modeling and scoring GIs, however, capture a continuous spectrum of phenotype strengths (Fig. 1B; Schuldiner et al. 2005; Collins et al. 2006; Baryshnikova et al. 2010; Collins et al. 2010; Horn et al. 2011) and are normally centered at zero (i.e., a noninteracting gene pair). For example, the E-MAP approach (epistasis miniarray profile) uses the SGA platform for high-throughput generation of double mutants and a continuous genetic interaction score (S-score), capturing the full spectrum of interaction strengths for discovery of both positive and negative GIs (Collins et al. 2006).
COMPUTATIONAL ANALYSIS OF EPISTATIC MAPS
High-throughput epistasis mapping measures and provides quantitative scores for thousands of interactions, each of which may be difficult to interpret in isolation. Hence, computational tools are required to analyze and visualize the resulting data sets. These include methods that rely on genetic interactions alone (Collins et al. 2007; Pu et al. 2008; Jaimovich et al. 2010) and those that integrate additional data, such as protein–protein interactions (Ulitsky et al. 2008; Srivas et al. 2011) or enzyme–substrate relationships (Fiedler et al. 2009). The most broadly used approach for analyzing quantitative genetic interactions, and perhaps the most intuitive, is the visualization of heat maps created by hierarchical clustering. In this approach, the “genetic interaction profile” of a mutant (i.e., the set of GI scores associated with that mutant) is used as a high-dimensional phenotype. Mutants with similar genetic interaction profiles are grouped together (Fig. 1C). Mutants that perturb the same pathway or complex typically display similar genetic interaction profiles (Tong et al. 2004; Schuldiner et al. 2005; Ye et al. 2005; Roguev et al. 2008; Costanzo et al. 2010; Frost et al. 2012; Ryan et al. 2012), and thus the clustering presents an unbiased view of pathway organization. In recent years, this approach has led to the identification of new members of well-characterized pathways (e.g., rsh1, a new component of the S. pombe RNAi machinery [Roguev et al. 2008]), unbiased functional annotation of more than 150 S. pombe genes, and annotation of more than 300 functional modules (Ryan et al. 2012). Additionally, comparative analysis of epistasis maps from budding and fission yeast has revealed some of the principles of GI network evolution and functional module repurposing (Roguev et al. 2008; Frost et al. 2012; Ryan et al. 2012).
HIGH-THROUGHPUT EPISTASIS MAPPING IN S. pombe
Inspired by the host of biological insights gained from systematic epistasis mapping in S. cerevisiae, significant effort has been directed in recent years toward the development of methods for quantitative genetic interaction mapping in S. pombe. Two key ingredients contributed to the success of this effort—the construction of a genome-wide deletion library in S. pombe (Kim et al. 2010) and the development of two experimental platforms for rapid generation of double mutants: the PEM system (Pombe Epistasis Mapper) (Roguev et al. 2007) and the SpSGA method (Dixon et al. 2008).
The S. pombe deletion library is available in two flavors—a heterozygous diploid set encompassing 4836 deletions (98.4% of the genome in which one gene has been assigned as being 100 amino acids or larger) and a haploid set containing 3400 deletions of nonessential genes (95.3% of the nonessential genome in which one gene has been assigned as being 100 amino acids or larger). This resource has been successfully used in a number of chemogenomic (Kennedy et al. 2008; Fang et al. 2012; Zhou et al. 2013; Rallis et al. 2014) and functional genomics (Deshpande et al. 2009; Pan et al. 2012; Tange et al. 2012; Hayles et al. 2013) screens.
Although differing in implementation, both the PEM and SpSGA methods apply a series of selection steps to generate and isolate haploid double mutant species for subsequent analysis. Both systems are amenable to automation and have been used successfully for generation of quantitative epistasis maps in S. pombe; however, the bulk of the available genetic interaction data in this organism have been generated using the PEM system (Roguev et al. 2008; Kim et al. 2009, 2014; Wiseman et al. 2009; Stewart et al. 2011; Frost et al. 2012; Ryan et al. 2012; Sanchez et al. 2012; Kabeche et al. 2014; Kriegenburg et al. 2014), which is discussed in more detail below.
The PEM system (Fig. 2; Roguev et al. 2007) is designed to facilitate high-throughput generation of haploid double mutants in S. pombe and follows the logic behind the original SGA platform (Tong et al. 2001). Haploid double mutants are created on agar plates through mating and a series of selection steps. First, heterozygous diploids are generated by allowing a query strain containing a particular gene deletion or mutation to mate with a library of gene deletions. Subsequent meiosis and sporulation produces a complex cell mixture that includes unmated parental cells, nonsporulated diploids, and the randomly assorted haploid meiotic products. To study the phenotype of the double mutants, it is therefore critical to eliminate all contaminating species. To this end, three key selections are implemented in the PEM system: (i) antidiploid selection (ADS), which eliminates remaining diploid cells; (ii) mating-type selection (MTS), which, by eliminating one of the mating types, generates a uniform population of cells of only one mating type, prevents remating, and makes it possible to easily perform another genetic cross if needed; and (iii) a double-mutant selection (DMS), which eliminates wild-type and single-mutant haploid species.
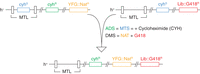
A schematic representation of a genetic cross using the PEM system. cyhS and cyhR are cycloheximide (CYH)-sensitive (rpl42+) and -resistant (rpl42-P56Q) alleles, respectively. YFG::NAT and Lib::G418 are query and library deletions, respectively. MTL, mating-type locus; ADS, antidiploid selection; MTS, mating-type selection; DMS, double-mutant selection.
The selectable markers for ADS and MTS are introduced on the query side of the genetic cross, thus making the PEM system independent of the genetic background of the library. The ADS strategy in the PEM system makes use of the protein synthesis inhibitor cycloheximide and a recessive mutation (P56Q) in a large ribosomal subunit protein L36/L42 encoded by rpl42 (SPAC15E1.03). The wild-type allele of rpl42 (rpl42+) renders S. pombe naturally sensitive to cycloheximide, whereas the P56Q allele (rpl42-P56Q) confers a robust resistance to the drug in a haploid context without causing apparent growth defects (Roguev et al. 2007). After mating, the resulting diploids are heterozygous for rpl42 (with rpl42-P56Q and rpl42+ coming from the query and library sides of the cross, respectively); therefore, they are cycloheximide-sensitive and eliminated on treatment with cycloheximide. MTS relies on introducing a copy of rpl42+ in the mating-type locus of the query strain. This allows for cycloheximide-based counterselection of this mating type, thus generating a uniform population of h+ haploids and preventing remating within the same cell pool. In the procedures outlined in Protocol: Genetic Interaction Screens in Schizosaccharomyces pombe Using the Pombe Epistasis Mapper (PEM) System and a Manual Colony Replicator (Colson and Hartsuiker 2016) and Protocol: Genetic Interaction Mapping in Schizosaccharomyces pombe Using the Pombe Epistasis Mapper (PEM) System and a ROTOR HDA Colony Replicating Robot in a 1536 Array Format (Roguev et al. 2016a), ADS and MTS are performed simultaneously, making the protocols faster to execute and more cost-efficient. Additionally, because all resulting haploids are from the same mating type, it is possible to easily perform consecutive genetic crosses and generate triple- and higher-order mutants to dissect more complex pathways with higher degrees of redundancy (Haber et al. 2013; Braberg et al. 2014).
DMS is usually achieved using two dominant antibiotic resistance markers, KanMX6 and NatMX6, which confer resistance to geneticin (G418) and nourseothricin (NAT), respectively. It can also be performed using auxotrophic markers. The use of antibiotic resistance markers instead of auxotrophic mutations is advantageous, as screens can be performed on relatively inexpensive rich media. It also makes the protocol faster and independent of the genetic background of the library.
The SpSGA method (Dixon et al. 2008) starts with the mating of haploid cells of opposite mating types on minimal medium. The resulting diploids are allowed to undergo meiosis and sporulation. ADS in this method relies on enrichment for meiotic products by incubation at 42°C, which removes unmated parental haploid and unsporulated diploid cells. Surviving spores are then allowed to germinate on rich medium, but, in contrast to the PEM system, no MTS selection is applied. This results in a mixed population of h+ and h− cells. Double mutants are subsequently selected using dominant antibiotic resistance markers similar to those used in the PEM system.
Because both the PEM and SpSGA systems are, in essence, methods for rapid generation of haploid genotypes through a genetic cross, their utility is not limited to large-scale genetic interaction mapping. For example, “crossing in” a point mutant or a temperature-sensitive or affinity/fluorescently tagged allele of the gene of interest can be performed with ease as long as these can be selected for (e.g., by positioning a selectable antibiotic or auxotrophic marker in close proximity to the gene of interest).
Manual replication provides a good, low-cost alternative for genetic interaction screening for laboratories in which a colony replicating robot is not available (see Protocol: Genetic Interaction Screens in Schizosaccharomyces pombe Using the Pombe Epistasis Mapping (PEM) System and a Manual Colony Replicator [Colson and Hartsuiker 2016]). The effect of genotype and/or the environmental impact on the growth of single and double mutants is typically quantified after imaging the colony arrays. Imaging can be performed at regular intervals to allow the construction of growth curves (Lawless et al. 2010; Banks et al. 2012), or fitness analysis can be based on a single image obtained at the end of the experiment (i.e., end point analysis); see Protocol: Genetic Interaction Score (S-Score) Calculation, Clustering, and Visualization of Genetic Interaction Profiles for Yeast (Roguev et al. 2016b). Growth curve analysis through calculation of growth parameters offers certain advantages, as it provides a more information-rich and potentially more accurate measure of fitness, including doubling time, lag, exponential, and stationary/starvation phase duration. However, it generally requires some degree of laboratory automation and its throughput is not as high as screening methods based on end point measurement (Banks et al. 2012).
ACKNOWLEDGMENTS
This work is supported by National Institutes of Health grant R01 GM084279.
Footnotes
-
From the Fission Yeast collection, edited by Iain M. Hagan, Antony M. Carr, Agnes Grallert, and Paul Nurse.








