
Single-Cell and Spatial Transcriptomic Analysis of Maize Embryo Development
- School of Integrative Plant Science, Plant Biology Section, Cornell University, Ithaca, New York 14853, USA
- ↵2Correspondence: haowu{at}njau.edu.cn
Abstract
Plant embryogenesis encompasses the biological processes wherein the zygote (fertilized egg) undergoes cell division, cell expansion, and cell differentiation to develop histological tissue layers, meristems, and various organs comprising the primordial body plan of the organism. Studies of embryogenesis in the agronomically important maize crop advance our understanding of the fundamental mechanism of plant development, which, upon translation, may advance agronomic improvement, optimization of conditions for somatic embryogenesis, and plant synthetic biology. Maize embryo development is coordinated temporally and spatially and is regulated by interactive genetic networks. Single-cell RNA sequencing (RNA-seq) and spatial transcriptomics are powerful tools to examine gene expression patterns and regulatory networks at single-cell resolution and in a spatial context, respectively. Single-cell technology enables profiling of three-dimensional samples with high cellular resolution, but it can be difficult to identify specific cell clusters due to a lack of known markers in most plant species. In contrast, spatial transcriptomics provide transcriptomic profiling of discrete regions within a sectioned, two-dimensional sample, although single-cell resolution is typically not obtained and fewer transcripts per cell are detected than in single-cell RNA-seq. In this review, we describe the combined use of these two transcriptomic strategies to study maize embryogenesis with synergistic results.
INTRODUCTION
Maize (Zea mays) is a widely cultivated cereal crop with an overall annual yield of more than 1161.86 million metric tons (http://www.worldagriculturalproduction.com/). Its significance in agricultural and industrial applications is well described (Strable and Scanlon 2009). Maize embryo development comprises the dynamic, spatial–temporal formation of the first tissues and organs of the plant, although in plants, embryogenesis continues throughout the life cycle. As shown in Figure 1, after fertilization, the zygote undergoes an asymmetric division to form a small apical cell and a large basal cell, which develop into the embryo proper and suspensor, respectively. The embryo proper establishes both the shoot and root apices, which mediate all subsequent grass development. The nonpersistent suspensor is a supporting structure that anchors the embryo proper in the endosperm cavity, and enables transfer of nutrients and metabolites to early-staged embryos; at maturity, the suspensor is fully degraded (Steeves and Sussex 1989). The first embryonic organs formed from the maize embryo proper are the shoot apical meristem (SAM) and the scutellum. The SAM is a pool of stem cells that is ultimately responsible for the development of all the organs in the adult shoot. The scutellum is the first-emerged embryonic lateral organ, which serves specialized digestive functions during endosperm digestion and nutrient transfer in the germinating seedling. Subsequently, the coleoptile initiates as the second-emerged embryonic lateral organ, arising from the apical SAM periphery and forming a fused, cylindrical leaf-like organ that entirely encloses the shoot apex. One function of the sheathing coleoptile is to protect the enclosed, underlying seedling (i.e., the SAM and four to five leaf primordia) during germination. The third lateral organ to emerge in the grass embryo is the first foliar leaf (L1), which initiates on the germinal side of the SAM. Mature seeds of most maize inbred lines contain four to five juvenile leaf primordia, all initiated during embryogenesis in the distichous phyllotactic pattern, where the leaves are arranged in two alternate sides on a stem (Randolph 1936; Abbe and Stein 1954; Poethig et al. 1986; Bommert and Werr 2001; Nardmann and Werr 2009).
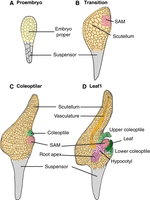
General overview of maize embryogenesis until the Leaf1 stage. (A) Proembryo stage. After fertilization, the zygote undergoes asymmetric cell division to form a small apical cell and a larger basal cell, which develop into the embryo proper and suspensor, respectively. (B) Transition stage. The embryo proper cells differentiate into a shoot apical meristem (SAM) on the embryo's germinal face and the scutellum on the abgerminal side. (C) Coleoptilar stage. The coleoptile initiates above the SAM adjacent to the scutellum. (D) Leaf1 stage. The first leaf primordium initiates from the SAM. Cells below the SAM differentiate into the hypocotyl and root apex. Vasculature tissue becomes histologically visible in the scutellum. The figure was adapted with permission from Wu et al. (2024) under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
The ontology and homology of the grass cotyledon have been subjects of botanical debate for over a century. One model proposes that the scutellum comprises the cotyledon, whereas the coleoptile is a modified foliar leaf (Weatherwax 1920). Another model suggests that the coleoptile is the cotyledon, and the scutellum is an evolutionary innovation of the grasses (Boyd 1931). Kaplan (1996) and others proposed a bipartite model wherein the scutellum and coleoptile comprise the distal and proximal domains of the single grass cotyledon, respectively. Analyses of the first three embryonic lateral organs generated in the grass embryo provide insight into discussions on the controversial evolutionary–developmental (evo-devo) biology of the grass cotyledon, and may provide insights into the ontogeny and homology of grass embryonic organs.
Expression of genes implicated in cell differentiation and patterning vary significantly across discrete maize embryonic tissues and organs (Takacs et al. 2012). Previous protocols used in comparative transcriptomic analyses of discrete tissues and organs include fluorescence-activated cell sorting (FACS) and laser microdissection RNA-seq (LM-RNA-seq). The relative scarcity of transgenic, fluorescent reporter lines has limited the application of FACS in grasses. LM-RNA-seq technology enables precise dissection and collection of specific, microscopic cell types and tissues for gene expression profiling, but does not provide single-cell resolution. Modern tools such as single-cell RNA-seq (scRNA-seq) and spatial transcriptomic analyses overcome these limitations and reveal cell-specific patterns of gene expression at high-throughput and -resolution. In this review, we explore the strengths and weaknesses of each approach and discuss their use in assaying gene expression in maize embryogenesis. In our associated protocol, we provide detailed instructions for carrying out both scRNA-seq and spatial transcriptomic analyses on maize embryos (Wu and Scanlon 2025).
Single-Cell Transcriptomic Analyses
Single-cell transcriptomic analysis, often referred to as scRNA-seq, is a cutting-edge technology that profiles gene expression at single-cell resolution. The scRNA-seq protocol comprises five major steps (Fig. 2): (1) isolation of cells (protoplasts), (2) single-cell barcoding, (3) library construction, (4) RNA sequencing, and (5) data processing and analysis (Yu et al. 2023). For plants, the isolation of individual cells requires removal of the plant cell wall and complete dissociation of the resulting protoplasts from undigested plant cells/tissues, using a combination of cell wall-digesting enzymes. The protoplast suspension is loaded into the 10x Genomics Chromium Platform to generate a Gel Bead-In-EMulsion (GEM) (10x Genomics, Inc. 2025). The GEM enables the encapsulation of individual protoplasts in microscopic aqueous droplets (microfluidics beads) along with a gel bead containing unique barcoded primers. RNA within each protoplast is captured on the beads, whereafter barcoded cDNA libraries are built following reverse transcription. Each cell-specific cDNA library is then sent for massively parallel sequencing, and the resulting data are processed to enable cell clustering and cell differentiation trajectory projections (pseudotime studies). In single-cell clustering analyses, most of the significant patterns of variation (i.e., most informative genes) in the data are captured via dimensionality reduction approaches, such as principal component analysis (PCA), followed by visualization of the clusters by uniform manifold approximation and projection (UMAP). PCA is a linear transformation method that projects high-dimensional data onto a smaller set of uncorrelated principal components (PCs). The first few PCs capture most of the variance in the data set, helping to identify major patterns in cell-to-cell variation. UMAP is a nonlinear technique that projects high-dimensional data into a lower-dimensional space (usually 2D or 3D) while preserving local relationships between cells (McInnes et al. 2018; Tsuyuzaki et al. 2020). In cell differentiation trajectory projections or pseudotime studies (Hou et al. 2023), the algorithms use reduced-dimensional data to model the continuous changes in gene expression across cells and then reconstruct the trajectory of the underlying biological process. Once the trajectory is reconstructed, each cell is assigned a pseudotime value. This value represents the position of the cell along the trajectory and estimates its relative stage in the biological process.
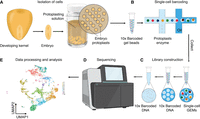
A flow chart for performing single-cell transcriptomic analysis. (A) Maize embryos are dissected from developing kernels. Cell walls are digested in digesting solution, followed by isolation of the resulting protoplasts. (B) Protoplasts are mixed with 10x barcoded gel beads and embedded in oil, forming a Gel Bead-In-EMulsion (GEM). (C) Single-cell GEMs are collected; cDNA is synthesized through reverse transcription (RT). cDNA strands are then pooled, followed by oil removal. The cDNA molecules are amplified for library construction. (D) The cDNA library is sent to be sequenced. (E) The read data are processed for subsequent analyses. The example graph shows a uniform manifold approximation and projection (UMAP) layout with 11 cell clusters (clusters 0–10 coded by different colors). Steps B and C are adapted from the 10x Genomics User Guide: CG000732 (10x Genomics, Inc. 2024a). Images courtesy of 10x Genomics, Inc.
The Arabidopsis root was among the first and most-studied plant structures using scRNA-seq technology. In 2016, individual Arabidopsis root cell protoplasts were first used to study stem cell regeneration using scRNA-seq (Efroni et al. 2016). Several subsequent studies focused on Arabidopsis root cell types and root developmental trajectory/pseudotime analyses (Denyer et al. 2019; Zhang et al. 2019), abiotic stress responses (Jean-Baptiste et al. 2019), xylem cell differentiation (Turco et al. 2019), and lateral root formation (Gala et al. 2021; Serrano-Ron et al. 2021).
In the first application of scRNA-seq analysis in grasses, Nelms and Walbot (2019) characterized gene expression programs in single cells comprising the premeiotic stages of male germline development in maize. The study found that nearly all pregerminal cells proliferate, which fails to support a stem cell model of generating meiotic cells. Subsequently, Satterlee et al. (2020) profiled the transcriptome and inferred the cell differentiation trajectory of the maize shoot apex at single-cell resolution. Pseudotime analyses revealed that ectopic expression of KNOTTED1 (KN1) accelerates cell differentiation in maize leaves by promoting sheath development. Subsequently, Liu et al. (2021) profiled the single-cell transcriptome of root tips in rice, and identified novel cell types associated with phytohormone biosynthesis, signaling, and response. Later scRNA-seq profiled cell-specific gene expression and differentiation of additional grass organs, including maize and Setaria roots (Ortiz-Ramírez et al. 2021), leaves and roots in rice (Wang et al. 2021; Zhang et al. 2021), the maize female inflorescence (Xu et al. 2021), the rice flower (Zong et al. 2022), and maize seedling leaves (Satterlee et al. 2023). These studies demonstrate how the application of scRNA-seq technology can deliver broad insights into plant development and differentiation via the detection of novel cell types and the construction of gene expression landscapes across diverse tissue types. Moreover, scRNA-seq facilitates the tracking of single-cell differentiation over developmental time via pseudotime analyses that are not possible using conventional, bulk RNA-seq.
Spatial Transcriptomic Analyses
One drawback to conventional scRNA-seq analyses of plants is that the protoplasting technology removes cells from their spatial context within the plant. Consequently, prior (or subsequent) studies of cell/tissue-specific gene expression are required to identify the original, in planta location of each cell analyzed in a scRNA-seq experiment. In contrast, spatial transcriptomics enable gene expression profiling while preserving the spatial context of the cells, tissues, and organs analyzed. Two major strategies for spatial transcriptomic methods are currently available: fluorescence in situ hybridization (FISH)-based and high-throughput sequencing-based studies. FISH-based spatial transcriptomic approaches use multiplex fluorescence in situ hybridization (M-FISH) techniques, wherein multiple gene-specific probes using different fluorophores bind to the native transcripts within cells/tissue and enable expression analyses of multiple genes simultaneously (Moffitt et al. 2016). Although M-FISH protocols are commercially available (Laureyns et al. 2022), high-throughput analyses are currently unavailable and thus will not be further discussed in this short review. Sequence-based spatial transcriptomic protocols use universal poly(dT) probes to capture mRNA transcripts that are subsequently reverse-transcribed, amplified, and sequenced, while separate spatial barcodes identify the locations of these transcripts (Stahl et al. 2016). Specifically, spatial barcodes comprise specific probe sequences for every capture spot on the sample slide, thereby delivering spatial information for each transcript detected. For example, if an mRNA transcript for Gene X is detected with Barcode #A123, we know it came from Spot A123 on the tissue. Likewise, if Barcode #B456 contains transcripts for Gene Y, we know it originated from Spot B456. By combining sequencing data with the barcode map, a spatial gene expression heat map is generated, showing where different genes are expressed in the tissue.
High-throughput sequencing-based spatial transcriptomic methods feature five major steps (Fig. 3): (1) sample collection and histological sectioning; (2) tissue optimization (optional), tissue permeabilization, and RNA binding to probes; (3) library construction; (4) RNA sequencing; and (5) data processing and analysis. Most current spatial transcriptomic platforms, such as 10x Genomics Visium, Slide-seq, and Stereo-seq, require fresh-frozen samples embedded in an optimal cutting temperature (OCT) reagent to maintain RNA quality, whereafter samples are cryosectioned, slide-mounted, and stained (Rodriques et al. 2019; Fawkner-Corbett et al. 2021; Wei et al. 2022). Sample slides are preprinted with multiple universal probes containing spatial barcode information as well as unique molecular identifiers (UMIs), thereby yielding spatial information while also labeling every individual, captured RNA molecule. Probes may be allotted into RNA capture spot arrays or bound to beads immobilized on the slides; probes on each individual capture spot (or bead) share the same spatial barcodes. Tissues are then permeabilized to release RNA molecules from the tissue sections and mRNAs hybridize to the underlying probes within the beads/spots. Tissue permeabilization time is a critical parameter affecting data quality. If the permeabilization time is too short, insufficient amounts of RNA may be released from the tissue, causing data loss. In contrast, if the permeabilization time is too long, the released RNA may diffuse to adjacent geographic locations, resulting in errant spatial information. As such, the tissue permeabilization time must be optimized when processing any tissue/organ/species for the first time. The released RNA is then reverse-transcribed into cDNA, and amplified prior to library construction. Because the slide-bound probes are used for reverse transcription, each cDNA molecule contains a spatial barcode and a UMI. Resultant libraries are then sent for sequencing, and the resulting data are mapped to the corresponding geographic location of the tissue sections using spatial barcode data. In this way, gene expression data are correlated to geographic locations containing specific cells/tissues/organs.
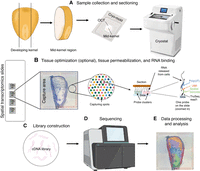
A flow chart for performing spatial transcriptomic analysis. (A) The developing maize kernel is cut longitudinally, and the mid-kernel region containing the embryo is retained. The mid-kernel region is fresh-frozen in optimal cutting temperature (OCT) and sectioned using a cryostat. (B) Tissue sections are mounted on the capture areas of spatial transcriptomic slides. During tissue permeabilization, RNA is released from the section and binds to probes on capture spots. Universal poly(dT) probes contain spatial barcode information and unique molecular identifiers (UMIs). When processing a tissue or organ for the first time, we highly recommend performing a tissue optimization step to determine the optimal permeabilization time. If the permeabilization time is too short, the tissue remains intact, leading to incomplete RNA release, resulting in low transcript detection and poor gene expression signals, while if too long, it may cause RNA diffusion, leading to mislocalization of transcripts and reduced spatial specificity. (C) First-strand cDNA is synthesized via reverse transcription (RT), followed by second-strand cDNA synthesis. The double-stranded cDNA molecules are then released from the slides and undergo amplification for library construction. (D) The cDNA library is sent to be sequenced. (E) The read data are processed and mapped to the sections based on the spatial barcode. Step B is adapted from the Visium Spatial Gene Expression User Guide CG000239 (10x Genomics, Inc. 2024b). Images courtesy of 10x Genomics, Inc.
The Visium spatial transcriptomic platform is most often used in animal studies (Tirado-Lee 2021), but was first used in plants in studies of Arabidopsis inflorescence meristems, leaf buds in Populus tremula, and female cone development in Picea abies (Giacomello et al. 2017; Giacomello and Lundeberg 2018). A modified Visium protocol was used in our recent, spatial transcriptomic study of maize embryo development (Wu et al. 2025). Another newly emerged spatial transcriptomic protocol termed “Stereo-seq” combines nanoball array chips (chips on a slide coated with densely packed probe nanoballs that carry spatial barcodes) and in situ RNA capture to achieve transcriptomic analyses at subcellular-level resolution (Liu et al. 2022; Xia et al. 2022).
Several features of plant cells present challenges to their study via spatial transcriptomics. Many plant cells contain large, water-filled vacuoles that readily form ice crystals upon freezing; when cryosectioned, these ice-filled vacuoles may destroy both subcellular structures and tissue integrity. Likewise, the cell wall inhibits cell permeabilization, which limits the ability of RNA molecules to interact with the underlying slide bead, such that ultra-thin tissue sections (i.e., less than one cell diameter thick) or cell wall digestion may be required to enable tissue permeabilization. Once these challenges are overcome, spatial transcriptomics are powerful tools providing in situ transcriptomic information for plant cells.
Integration of Single-Cell RNA Sequencing and Spatial Transcriptomic Approaches
Although both single-cell RNA sequencing and spatial transcriptomics are cutting-edge technologies used to identify specific cell clusters and infer cell differentiation trajectories in plant tissues, each technique has advantages and limitations that are summarized in Table 1. For example, scRNA-seq profiles the transcriptomes of three-dimensional samples at single-cell resolution and may detect several thousand gene transcripts for each of several thousand cells (Satterlee et al. 2020, 2023; Shahan et al. 2022). Such large data sets boast statistical power, data generalizability, and reduced sample bias, all of which aid in the identification of rare cell types and low level transcripts. However, scRNA-seq does present complications specific to plant cells. The process of cell wall digestion may reduce the viability of plant cells and increase the risk of RNA degradation. Plant tissues contain cells of variable sizes and cell wall architectures, which may introduce cell-type biases after protoplast digestion. For instance, some plant cells contain secondary cell walls that may hinder protoplast digestion, whereas extra-large cells may block the microfluidic processes generating the GEM, resulting in the absence of certain cell types from the data sets. In such cases, researchers may use single-nucleus RNA-seq (snRNA-seq) as an alternative, although nuclei typically yield far less RNA than a whole protoplast (Bakken et al. 2018; Selewa et al. 2020; Chen et al. 2023). In addition, many cell/tissue-specific marker genes are unavailable for many plant species, presenting problems in identification of the tissue-specific origins of cell clusters. In such cases, researchers must conduct a series of time-consuming and labor-intensive analyses to identify cell cluster–specific marker genes.
Advantages and limitations of the single-cell and spatial transcriptomic approaches
Spatial transcriptomic approaches partially compensate for the limitations of scRNA-seq procedures. Cryosectioning preserves RNA quality, thereby minimizing the risk of RNA degradation. Moreover, spatial transcriptomic approaches present fewer limitations on cell size, although this may be a factor influencing RNA migration. Cell wall properties may likewise hinder RNA diffusion, although adjusting the thickness of the cryosectioned samples may reduce this problem. Critically, spatial transcriptomic approaches yield gene expression data while preserving the geospatial context, thereby assisting the identification of cell clusters.
Among the noted limitations of spatial transcriptomic methods is the so-called resolution gene detection dilemma. Giacomello et al. (2017) used spatial transcriptomic slides featuring capture spots of 100 µm diameter, where the center-to-center distance was 200 µm (i.e., a 100 µm gap between two adjacent spots). Maize shoot meristematic cells are ∼10 µm in diameter, such that the capture spot area and the gap area may include 10 or more cells. This relatively low resolution may reduce the efficiency of spatially locating transcripts from smaller cells/tissues and young organ primordia. Recently, 10x Visium doubled the bead resolution of their spatial transcriptomic slide platform, although the system still struggles to parse signals from closely packed tissues or organs. For example, the maize embryonic SAM, coleoptile, and leaf are closely positioned within a 150–200 µm circle area, so that a Visium capture spot may encompass multiple organs (Fig. 4). Slide-seq and Stereo-seq protocols feature improved resolution of up to 10 µm and 500 nm, respectively, thereby exceeding single-cellular or subcellular resolution (Yu et al. 2023). However, one drawback to this higher resolution is a dramatic reduction in the sensitivity of transcript detection, defined as the percentage of detected unique transcripts relative to the total number of unique transcripts in a particular area (Stahl et al. 2016; Asp et al. 2020). BGI Genomics asserts that Stereo-seq is able to detect greater than 500 genes per cell (https://www.bgi.com/global), although no demonstrations have been reported in plant research.
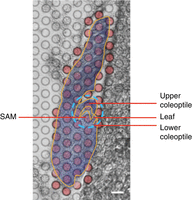
Maize embryo overlaid with the Visium capture spots. The red circles are Visium capture spots covered by the embryo section. The shoot apical meristem (SAM), upper and lower coleoptile, and leaf are located within the blue-circled area. Scale bar, 100 µm.
A second limitation is that data gathered from spatial transcriptomic approaches may fail to represent the entire, three-dimensional sample of interest. Cells may not be distributed or arranged homogeneously within a tissue sample, and different sectional planes may reveal distinct cell-type profiles. For example, a transverse section through the maize embryo may visualize the SAM but can miss the root pole. Therefore, it may be necessary to process multiple sections from different planar axes in order to sample cells from these different tissues. The application of spatial transcriptomic protocols in high throughput, on complete serial sections of three-dimensional samples, is not yet practical.
CONCLUSION
Owing to their relatively small vacuoles and thin cell walls, maize embryonic cells are ideal for single-cell RNA-seq and spatial transcriptomic research. To overcome the specific limitations currently inherent to each of these two protocols, we combined the two analyses in an integrated approach that enhances our understanding of the spatial organization and expression landscapes of individual cells in this complex biological system (Wu and Scanlon 2025, in this collection; Wu et al. 2025). In this review, we suggest that scRNA-seq analyses provide the majority of the cell-specific transcriptomic data, whereas the spatial transcriptomic procedures are critical to the verification of cell cluster identities. When selecting from among the variety of currently available spatial transcriptomic protocols, researchers must strike a balance between limitations in gene detection and spatial resolution.
COMPETING INTEREST STATEMENT
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Investigation: H.W. Visualization: H.W. Writing—original draft: H.W. Writing—review and editing: M.J.S.
Footnotes
-
From the Maize collection, edited by Candice N. Hirsch and Marna D. Yandeau-Nelson. The entire Maize collection is available online at Cold Spring Harbor Protocols and can be accessed at https://cshprotocols.cshlp.org/.
REFERENCES
*Reference is also in this subject collection.








