
Whole-Mount RNA In Situ Hybridization and Immunofluorescence of Xenopus Embryos and Tadpoles
- Department of Psychiatry and Behavioral Sciences, Weill Institute for Neurosciences, University of California, San Francisco, San Francisco, California 94143, USA
- ↵1Correspondence: helen.willsey{at}ucsf.edu
Abstract
A major advantage of experimentation in Xenopus is the ability to query the localization of endogenous proteins and RNAs in situ in the entire animal during all of development. Here I describe three variations of staining to visualize mRNAs and proteins in developing Xenopus embryos and tadpoles. The first section outlines a traditional colorimetric staining for mRNAs that is suitable for all stages of development, and the second extends this protocol for fluorescence-based detection for higher spatial and quantitative resolution. The final section details detection of proteins by immunofluorescence, optimized for tadpole stages but widely applicable to others. Finally, optimization strategies are provided.
MATERIALS
Reagents
Reagents required for the colorimetric RNA in situ hybridization procedure only (Steps 1–34)
Acetic anhydride (Sigma-Aldrich 320102)
Alkaline phosphatase buffer with tetramisole hydrochloride
Anti-digoxygenin-AP antibody (Sigma-Aldrich 11093274910)
Bleaching solution for in situ hybridization
BM purple (Sigma-Aldrich 11442074001)
Bouin's fixative for in situ hybridization
Digoxygenin-11-UTP-labeled RNA probes, in-vitro-transcribed (see Protocol: Synthesis and Purification of Digoxigenin-Labeled RNA Probes for In Situ Hybridization [Sive et al. 2007a])
-
Both an experimental probe and a control probe (either sense-transcribed, or a probe with a known, very specific pattern) should be used.
Hybridization buffer for colorimetric in situ hybridization
Methanol (Fisher A4544)
Molten agarose (1%) (optional; see Step 33)
Paraformaldehyde (4% in PTw)
-
Dilute 20% paraformaldehyde to 4% in PTw on staining day.
PBS with Tween 20 (PTw)
-
Add 0.1% Tween 20 to 1× PBS. Store at room temperature.
Phosphate-buffered saline (PBS) (10×; pH 7.4)
Proteinase K (10 mg/mL; Fisher EO0491)
Proteinase K (10 µg/mL in PTw)
RNase A (20 µg/mL; Fisher EN0531)
RNase T1 (10 µg/mL; Fisher EN0542)
Sodium chloride-sodium citrate buffer (SSC; 20×)
Triethanolamine (0.1 m, pH 7-8; Sigma-Aldrich T1502)
Xenopus tadpoles or embryos of any stage
Reagents required for the fluorescent RNA in situ hybridization by hybridization chain reaction (HCR) procedure only (Steps 35–46)
Acetic anhydride (Sigma-Aldrich 320102)
HCR probes (custom-designed from Molecular Technologies)
HCR hairpins (standard-designed from Molecular Technologies)
Hybridization buffer for fluorescent in situ hybridization (30%)
Methanol (Fisher A4544)
Molten agarose (1%) (optional; see Step 46)
Paraformaldehyde (4% in PTw)
-
Dilute 20% paraformaldehyde to 4% in PTw on staining day.
-
Add 0.1% Tween 20 to 1× PBS. Store at room temperature.
Proteinase K (10 mg/mL; Fisher EO0491)
Proteinase K (10 µg/mL in PTw)
Triethanolamine (0.1 m, pH 7–8) (Sigma-Aldrich T1502)
Xenopus tadpoles or embryos of any stage
Reagents required for the immunofluorescence procedure only (Steps 47–58)
Bleaching solution for immunofluorescence
CAS-Block (10% in PBT)
CAS-Block (Invitrogen 00-8120)
Gentamicin (optional; see Step 57)
Molten agarose (1%) (optional; see Step 58)
Paraformaldehyde (4% in PBS)
-
Dilute 20% paraformaldehyde to 4% in PBS on staining day.
Paraformaldehyde (20%)
PBS with Triton X-100 (PBT)
-
Add 0.1% Triton X-100 to 1× PBS. Store at room temperature.
Primary antibodies
-
Potential primary antibodies are listed in Table 1.
Common antibodies compatible with this immunostaining procedure in stage 46 tadpoles along with relevant information
Secondary antibodies compatible with the primary antibodies (fluorescently conjugated)
Xenopus tadpoles
Equipment
Aluminum foil
Equipment for basket format (high-throughput sample processing) only
Baskets (Fig. 1A)
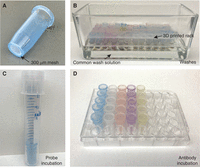
Basket format for higher-throughput staining. (A) Sample basket made from a 1.7-mL microcentrifuge tube (with cap and bottom cut off) with 300-µm mesh melted to the bottom. (B) Wash setup, with color-coded baskets arranged in a 3D-printed rack within a glass staining dish. Samples share a common wash buffer. (C) Probe incubation setup, in baskets within 15-mL round-bottom culture tubes with 500 µL of probe solution. (D) Antibody incubation setup within a 48-well culture plate with 500 µL of antibody solution in each well. These incubation setups allow for each sample to experience a different probe or antibody, if desired, and minimize the total volume required.
-
Construct the baskets from 1.7-mL tubes and 300-µm nylon mesh (Spectra Mesh 146487); for dissected tissue, use finer mesh. For basket-making instructions, see Protocol: Baskets for In Situ Hybridization and Immunohistochemistry (Sive et al. 2007b).
Basket racks (Fig. 1B)
-
Prepare the basket racks using 3D printer files available at willseyfroggers.org/resources. Basket racks can also be made by cutting the bottoms off of 1.7-mL tube racks.
Culture plates (48-well; Fisher 720086) (Fig. 1D)
Culture tubes (15-mL; Fisher 1496215E) (Fig. 1C)
Glass dish for washes (Fig. 1B)
-
Select a small dish (fits 24 baskets, holds 50 mL of wash buffer; Wheaton Inc. 900170), a medium dish (fits 30 baskets, holds 100 mL of wash buffer; Wheaton Inc. 900203), or a large dish (fits 60 baskets, holds 150 mL of wash buffer; Grainger 49WF37).
Lateral shaker at room temperature
Equipment for tube format (lower-throughput sample processing) only
Individual vials or tubes
Nutator at room temperature
Forceps
Glass slides and coverslips
Imaging stamps (Fig. 2A)

Imaging in agarose wells. (A) 3D-printed stamps for positioning embryos (left) or tadpoles (right). Agarose wells can be made by pressing these into molten agarose in a culture dish during cooling. (B) Tadpoles arranged into agarose wells. (C) Imaging animals using an upright stereoscope in agarose wells.
-
Prepare imaging stamps using 3D printer files available at willseyfroggers.org/resources.
Light box
-
Bright lights from a stereoscope can be substituted.
Syringes (1-mL)
-
These are used for dispensing vacuum grease onto glass slides for imaging.
Vacuum grease
Water bath (37°C) with lateral agitation for in situ RNAs
Water bath (60°C) with lateral agitation for in situ RNAs
Zeiss Axio Zoom.V16 microscope (or any appropriate microscope)
METHOD
-
Three separate procedures are described here: colorimetric whole-mount RNA in situ hybridization (Steps 1–34), fluorescence whole-mount RNA in situ hybridization by HCR (Steps 35–46), and whole-mount immunofluorescence (Steps 47–58). See the Discussion section to determine the best procedure for your experiment.
-
These procedures have been optimized for higher-throughput staining (24 to 60 samples processed in parallel), with each sample in a basket within a large rack in a glass staining dish sharing a common buffer solution (Fig. 1; see Protocol: Baskets for In Situ Hybridization and Immunohistochemistry [Sive et al. 2007b]). This protocol is also effective, although lower-throughput, in individual vials or tubes with manual washes.
Colorimetric Whole-Mount RNA In Situ Hybridization
-
Perform all steps on a lateral shaker with light agitation (∼40 rpm).
Fixation and Dehydration (2.5 h)
-
1. Fix animals for 2 h in 1× MEMFA solution at room temperature.
-
This step and the following one can be done in basket format or in individual vials or tubes if planning on long-term storage before staining. If using the basket format, limit the number of animals in a basket to 20 and make sure they are well-covered by solutions. Consult one's IACUC protocol for whether anesthetization is required before fixation.
-
-
2. Wash several times in methanol at room temperature to dehydrate. Freeze at least overnight at −20°C.
-
Samples can be stored long term at −20°C.
-
Rehydration and Permeabilization (55 min)
-
3. Rehydrate stepwise into PTw at room temperature as follows:
-
i. 5 min in 100% methanol,
-
ii. 5 min in 75% methanol and 25% H2O,
-
iii. 5 min in 50% methanol and 50% H2O, and
-
iv. 5 min in 25% methanol and 75% PTw.
-
-
4. Wash four times for 5 min each time in PTw at room temperature.
-
5. Permeabilize in 10 µg/mL proteinase K in PTw for 5 min at room temperature.
-
This step should be carefully monitored and not prolonged. For staining of superficial structures like epidermal cilia, omit this step. For staining of deeper structures, this step can be extended with careful testing or combined with dissection for further permeabilization.
-
Blocking and Hybridization (2.5 h for Xenopus tropicalis, 7.5 h for Xenopus laevis)
-
6. Wash twice for 5 min each time in 0.1 m triethanolamine (pH 7–8) at room temperature.
-
7. Wash twice for 5 min each time in 0.1 m triethanolamine with acetic anhydride (125 µL of acetic anhydride per 50 mL of 0.1 m triethanolamine) at room temperature.
-
8. Wash twice for 5 min each time in PTw at room temperature.
-
9. Refix for 20 min in 4% paraformaldehyde in PTw at room temperature.
-
10. Wash five times for 5 min each time in PTw at room temperature.
-
For fluorescence-based detection, proceed to Step 35.
-
-
11. Preheat probes at 1 µg/mL in hybridization buffer for colorimetric in situ hybridization for several hours at 60°C to help with penetration. In addition to your experimental probe, use a control probe (either sense-transcribed, or a probe with a known, very specific pattern).
-
12. Prehybridize samples in hybridization solution for 1 h (X. tropicalis) or 6 h (X. laevis) at 60°C with shaking.
-
Depending on the probe, this can be shortened to 1 h for X. laevis samples. Dissected tissues may also require less time than whole embryos.
-
-
13. Transfer samples into preheated probe solution overnight at 60°C with shaking.
-
Save the prehybridization solution to reuse the next day in Step 15. For the basket format, remove baskets from the rack and place into 15-mL round-bottom culture tubes with 500 µL of probe solution each (Fig. 1C). This allows each sample to have a different probe, if desired.
-
Probe Detection (Time Varies)
-
This is essentially an antibody staining against digoxygenin-11-UTP present in the RNA probe followed by enzymatic colorimetric detection. The procedure can be modified depending on the probe label and desired detection modality. Time to antibody incubation is 4.5 h; antibody incubation can be done overnight at 4°C or 4 h at room temperature; MAB washes can be done overnight at 4°C or for 5 h at room temperature; AP buffer washes take 15 min; and developing the stain in BM Purple varies from 1 h to days depending on the probe and sample.
-
14. Remove the probe solution and save at −20°C for reuse.
-
15. Wash samples in hybridization buffer, reused from Step 12, for 5 min at 60°C.
-
16. Wash twice for 3 min each time in 2× SSC at 60°C.
-
17. Wash three times for 20 min each time in 2× SSC at 60°C.
-
18. Incubate for 30 min at 37°C in 2× SSC containing 20 µg/mL RNase A and 10 µg/mL RNase T1.
-
19. Wash once for 10 min in 2× SSC at room temperature.
-
20. Wash twice for 30 min each time in 0.2× SSC at 60°C.
-
21. Wash twice for 10 min each time in 1× MAB at room temperature.
-
22. Incubate in 2% BMB blocking solution for at least 1 h at room temperature.
-
23. Incubate in antibody solution (dilute anti-digoxigenin-AP antibody 1:3000 in 2% BMB blocking solution) overnight at 4°C or for 4 h at room temperature.
-
For basket format in a dish, this is 16.6 μL of antibody per 50 mL of blocking solution. Alternatively, baskets can be transferred into a 48-well plate to use less total antibody, in which case each well has 500 μL of diluted antibody.
-
-
24. Wash five times for 1 h each time in 1× MAB at room temperature (or wash overnight at 4°C with multiple quick washes before and after overnight incubation).
-
25. Wash twice for 5 min each time in alkaline phosphatase buffer with tetramisole hydrochloride at room temperature.
-
26. Incubate in BM Purple reagent in wells of a 48-well plate at room temperature (Fig. 1D), protected from light with aluminum foil, and monitor until chromogenic reaction produces a stain of the desired intensity.
-
Incubation time in BM Purple varies widely depending on the probe and can only be determined empirically or by comparison to published literature for a given probe.
-
Depending on the stage and tissue interrogated, endogenous pigment may make the visualization of BM Purple precipitate difficult. Bleaching (see Steps 28–34) may make the signal easier to see. Consider this as the chromogenic reaction proceeds. If pigment precludes sensitive monitoring, consider using albino embryos at the start.
-
-
27. Stop the chromogenic reaction with a wash in 1× MAB at room temperature.
Postfixation and Bleaching (6 h)
-
28. Fix for at least 2 h in Bouin's fixative at room temperature.
-
29. Wash at room temperature in buffered ethanol solution 10 times for 10 min each time or until the embryos are no longer yellow.
-
30. Rehydrate stepwise into 1× SSC at room temperature as follows:
-
i. 5 min in 75% buffered ethanol and 25% 1× SSC,
-
ii. 5 min in 50% buffered ethanol and 50% 1× SSC,
-
iii. 5 min in 25% buffered ethanol and 75% 1× SSC, and
-
iv. twice for 5 min each time in 100% 1× SSC.
-
-
31. Bleach in bleaching solution for in situ hybridization for 1–2 h at room temperature under a light box or until embryos are white.
-
32. Wash three times for 5 min each time in 1× SSC at room temperature.
-
At this point, the samples are ready for imaging. The samples can be stored for years at −20°C in methanol or for months at 4°C in 1× SSC.
-
-
33. Mount the samples for imaging as follows:
-
For macroscale imaging, mount samples in agarose wells made using 3D printed stamps pressed in 1% molten agarose during cooling (Fig. 2).
-
3D printer files for stamps are available at willseyfroggers.org/resources.
-
-
Alternatively, mount on glass slides in 1× SSC within a vacuum grease well and affix a coverslip.
-
-
34. Image using brightfield microscopy.
-
See Troubleshooting.
-
Fluorescence Whole-Mount RNA In Situ Hybridization by Hybridization Chain Reaction (HCR)
-
This method is identical to the previous one until prehybridization. Perform all steps on a lateral shaker with light agitation (∼40 rpm).
Fixation, Dehydration, Rehydration, Permeabilization, and Blocking
-
35. Carry out Steps 1–10, and then proceed to Step 36.
Hybridization (35 min until Overnight Incubation; 1.5 h until Amplification)
-
36. Prehybridize samples in 30% probe hybridization buffer for fluorescent in situ hybridization for 30 min at 37°C.
-
37. Prepare probe solution by adding 2 pmol of each probe as provided by Molecular Technologies (1 μL of 2 μm stock per probe mixture) to 500 μL of 30% probe hybridization buffer that has been prewarmed to 37°C. In addition to your experimental probe, ideally use a control probe (either sense-transcribed, or a probe with a known, very specific pattern).
-
Probe volume can be reduced to the minimum required to cover samples.
-
-
38. Replace the 30% probe hybridization buffer with probe solution and incubate overnight (12–16 h) at 37°C with shaking.
-
For the basket format, remove baskets from the rack and place into 15-mL round bottom culture tubes with 300–500 µL of probe solution each (Fig. 1C).
-
-
39. Wash four times for 15 min each time in 30% probe wash buffer at 37°C with shaking.
-
Save probe solutions. They can be stored at −20°C and reused multiple times.
-
Heat wash solutions to 37°C before use.
-
-
40. Wash samples three times for 5 min each time in 5× SSCT at room temperature with shaking.
Amplification (35 min until Overnight Incubation; 1.5 h until Mounting)
-
41. Incubate samples in amplification buffer for 30 min at room temperature.
-
42. Prepare 30 pmol hairpin solutions (10 μL of each desired 3 μm hairpin) in amplification buffer as follows:
-
i. heat hairpins for 90 sec at 95°C,
-
ii. cool for 30 min in a dark drawer to room temperature, and
-
iii. add all hairpin solutions to amplification buffer (for a total volume of 500 µL) at room temperature.
-
-
43. Transfer samples into the hairpin solution and incubate overnight (12–16 h) in the dark at room temperature.
-
Hairpin solutions can be stored at −20°C and reused multiple times. For the basket format, remove baskets from the rack and place into 15-mL round bottom culture tubes with 300–500 µL of hairpin solution each (Fig. 1C).
-
-
44. Wash in 5× SSCT at room temperature as follows:
-
i. twice for 5 min each time,
-
ii. twice for 30 min each time, and
-
iii. once for 5 min.
-
-
45. Wash three times for 5 min each time in 1× SSC at room temperature.
-
At this point, the samples are ready for imaging. The samples can be stored for weeks in the dark at 4°C in 1× SSC.
-
-
46. Mount and image as follows:
-
For macroscale imaging, place in 1× SSC in agarose wells made using 3D printed stamps pressed in 1% molten agarose during cooling (Fig. 2). Image on an upright stereomicroscope with fluorescence.
-
3D printer files for stamps are available at willseyfroggers.org/resources.
-
-
For higher-magnification imaging, mount in 1× SSC in a vacuum grease well on a glass slide, affix coverslip, and image.
-
-
See Troubleshooting.
Whole-Mount Immunofluorescence
-
Perform all incubations (excluding antibody incubations) on a lateral shaker (basket format) or on a nutator (tube format).
Fixation (1 h)
-
47. Fix animals in 4% paraformaldehyde in PBS for 40 min at room temperature.
-
Consult one's IACUC protocol for whether anesthetization is required before fixation.
-
-
48. Wash in PBS three times for 5 min each time at room temperature.
Bleaching and Permeabilization (2 h, 5 min)
-
49. Bleach samples in bleaching solution for immunofluorescence for 1 h at room temperature under a light box.
-
This step is incompatible with phalloidin staining and will quench any fluorescent proteins (e.g., GFP); therefore, it should be omitted in those cases.
-
Bubbles are created in this step. If using tubes, transfer samples to a glass dish or open the tube tops to allow for gas release.
-
This step will remove pigmentation and provide some permeabilization. It should not be prolonged as it can begin to disintegrate the sample if performed for too long.
-
-
50. Permeabilize in PBT by washing three times for 20 min each time at room temperature.
-
For stages younger than 44, additional permeabilization may be required, such as dehydration.
-
Blocking and Incubation with Primary Antibody (1 h until Overnight Incubation)
-
51. Block in 10% CAS-Block in PBT for at least 1 h at room temperature.
-
52. Incubate in primary antibody diluted in 100% CAS-Block overnight at 4°C.
-
If using baskets, move the baskets into 48-well plates with 300 µL of antibody per well (Fig. 1D). If using tubes, use a minimum volume to cover animals completely.
-
A reasonable starting concentration for a new antibody is 1:100, but the concentration should be optimized empirically. For unconcentrated sera (e.g., from DSHB), start with a 1:5 dilution.
-
Washes and Incubation with Secondary Antibody (3 h, 10 min)
-
53. Wash in PBT three times for 10 min each time at room temperature.
-
54. Block in 10% CAS-Block in PBT for 30 min at room temperature.
-
55. Incubate in secondary antibody diluted in 100% CAS-Block for 2 h in the dark at room temperature.
-
If using baskets, move the baskets into 48-well plates with 300 µL of antibody per well (Fig. 1D). If using tubes, use a minimum volume to cover embryos completely.
-
If using fluorescence-conjugated secondary antibodies, cover tubes or baskets with aluminum foil to protect samples from the light for the remainder of the staining.
-
A typical commercial antibody dilution for this step is 1:250. Additional fluorescent dyes can be added during this step (e.g., DAPI).
-
Washes and Mounting (1.5 h until Mounting)
-
56. Wash three times for 10 min each time in PBT at room temperature.
-
57. Wash three times for 20 min each time in PBS at room temperature.
-
At this point, the samples are ready for imaging. The samples can be stored for a few weeks at 4°C in 1× PBS. If in solution, rather than mounted, add gentamicin (50 µg/mL) to the 1× PBS to extend storage time. An additional, final fixation for 40 min at room temperature in 4% paraformaldehyde in PBS (post-fixation) can also extend storage time if necessary.
-
-
58. Mount and image as follows:
-
For macroscale imaging, place in 1× PBS in agarose wells made using 3D printed stamps pressed in molten agarose during cooling (Fig. 2). Image on an upright stereomicroscope.
-
For higher-magnification imaging, mount in 1× PBS in a vacuum grease well on a glass slide, affix coverslip, and image.
-
See Troubleshooting.
-
TROUBLESHOOTING
Problem (Step 34, 46, or 58): Superficial staining is observed, but there is an absence of deeper tissue staining.
Solution: Increase permeabilization by a longer or more concentrated proteinase K treatment (for RNA hybridization), a longer or more concentrated detergent treatment (for RNA or protein staining), or by physically dissecting the tissue to expose the target region. Adding a dehydration step to the immunofluorescence protocol may increase permeabilization.
Problem (Step 34, 46, or 58): Tissue disintegrates during the procedure.
Solution: Increase fixation time and/or decrease proteinase K or detergent washes.
Problem (Step 34, 46, or 58): Excessive background staining is observed.
Solution: Increase stringency steps (longer 0.2× SSC washes and increased temperature for RNA hybridization; increased blocking time and permeabilization for immunostaining).
Problem (Step 58): Weak antibody staining is observed.
Solution: Empirically test alternative fixatives (e.g., try glutaraldehyde), antibody concentration, bleaching time, and/or detergent concentration.
Problem (Step 58): No signal is seen.
Solution: Refer to other excellent protocols for additional steps (dehydration, etc.), alternative fixatives, and additional positive control antibodies (e.g., Protocol: Whole-Mount Fluorescence Immunocytochemistry on Xenopus Embryos [Lee et al. 2008] and Brooks and Wallingford [2015]).
DISCUSSION
Three separate procedures are described in this protocol. The first involves whole-mount RNA in situ hybridization with colorimetric detection by BM Purple staining (Steps 1–34; Fig. 3A). This is a cost-effective strategy for assaying mRNA expression in embryos and tadpoles of all stages using in-vitro-transcribed digoxygenin-11-UTP-labeled RNA probes (see Protocol: Synthesis and Purification of Digoxigenin-Labeled RNA Probes for In Situ Hybridization [Sive et al. 2007a]). The second, whole-mount RNA in situ hybridization with fluorescent detection by HCR (Steps 35–46; Fig. 3B,C; Choi et al. 2018), is more expensive for assaying mRNA expression because it requires commercial RNA probes designed for Xenopus sequences (https://www.moleculartechnologies.org), but these probes can be reused. Fluorescence-based detection provides a great increase in spatial and quantitative resolution over colorimetric detection as well as the ability to label up to five RNAs in different wavelengths. Because of the nature of detection, there is also not the subjectivity of when to terminate the development of signal, which can be an advantage over colorimetric detection in some respects.
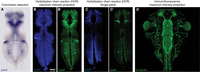
Sample RNA in situ hybridization and immunostaining micrographs. (A) Colorimetric staining for pax6 RNA (purple) in a stage 46 Xenopus tropicalis dissected brain imaged by widefield microscopy. (B,C) Fluorescence staining for pax6 RNA by HCR (green; B′, C′) costained with DAPI to label nuclei (blue; B, C) in a stage 46 X. tropicalis dissected brain imaged by confocal microscopy. (B–B′) Maximum intensity projection of confocal sections. (C–C′) Single imaging plane. Note the increased resolution potential with the fluorescence-based method. (D) Immunostaining for β-tubulin in the stage 46 X. tropicalis head region imaged by confocal microscopy.
The third procedure involves whole-mount immunostaining with fluorescent detection (Steps 47–58; Fig. 3D). Although it is optimized for tadpole stages, it also works well for many epitopes in earlier stages, including before gastrulation. See Table 1 for a list of primary antibodies compatible with this procedure, particularly for stage 46 tadpoles. Because this is one of the simpler procedures available, it is a good one to try first.
The procedures in this protocol should be modified according to the developmental stage and tissue type of interest. For example, dehydration is often helpful in earlier, more yolky stages, whereas it can interfere with staining in later tadpole stages. Some tissues and stages require physical permeabilization (e.g., removal of skin in later tadpole stages to better permeabilize the brain), whereas superficial tissues may need less permeabilization (e.g., omit the proteinase K step for epidermal cilia staining). Further, some antibodies produce better results with a particular fixative, permeabilization condition, etc., and require empirical testing to optimize (see Troubleshooting).
On a final note, the colorimetric RNA in situ procedure was derived from a widely used contribution from Joanna Yeh and Mustafa Khokha according to Sive et al. (2000) and originally described in Harland (1991). The fluorescent RNA in situ procedure was derived from Choi et al. (2018), and the imaging stamps were derived from Truchado-Garcia et al. (2018).
ACKNOWLEDGMENTS
I thank Cameron Exner for careful editing; Richard Harland, Edivinia Pangilinan, Mustafa Khokha, Maura Lane, Emily Mis, Karen Liu, Peter Walentek, Yuxiao Xu, and Cameron Exner for expert instruction and modifications of these procedures; Yuxiao Xu for the pax6 colorimetric image; and Albert Kim, Marta Truchado-Garcia, and Richard Harland for help with 3D-printing racks and stamps.








