
Advances in Phage Display—A Perspective
- ↵1Correspondence: smithgp194{at}gmail.com
Abstract
Phage display technology is enabled by genetic fusion of a foreign protein domain to a phage coat protein, without interfering with the phage's ability to replicate by infecting bacterial host cells. The displayed domain is exposed on the phage particle (virion) surface, where it can interact with molecules or other substances in the surrounding medium; in this regard, it acts like a normal protein. However, it possesses a superpower that is unavailable to ordinary proteins: It is easily replicated in great abundance because it is attached to a replicating virion whose genome includes its coding sequence. The main way this technology is exploited is construction of huge phage display “libraries,” comprising billions of phage clones, each displaying a different protein domain, and each represented by thousands, millions, or billions of genetically identical virions—all mixed together in a single vessel. Surface display allows exceedingly rare virions whose displayed protein domains happen to bind a user-defined molecule or other substance—generically called the “selector”—to be isolated from such libraries by an affinity selection process. The yield of selector-binding virions is much too low to be of practical use, but their number is readily increased by many orders of magnitude by propagating the virions in host bacteria in culture. This overview is a critical review of recent developments of this technology. It does not review the entire arena of contemporary phage display; there is special emphasis on phage display's most prominent application, phage antibodies, in which the displayed domain is an antibody domain, and the selector is an antigen of interest.
INTRODUCTION
The centerpiece of phage display is a type of engineered phage whose genome includes the coding sequence for a foreign protein domain genetically fused to one of the phage's coat protein genes. As a consequence of coat protein fusion, the foreign domain is exposed on the surface of the phage particle, called the virion, where it is accessible to molecules and other substances in the surrounding medium. The foreign domain is said to be “displayed” on the virion surface—hence the term “phage display.” In this sense, the displayed domain acts like an ordinary protein. However, at the same time, it can be replicated by many orders of magnitude, simply by infecting a culture of host bacteria with the virion that displays the domain and carries the domain's coding sequence in its genome.
Phage display's core processes are library construction and affinity selection. A typical phage display library contains billions of phage clones, displaying billions of different foreign domains on the virion surface. Affinity selection (explained briefly in the next section and more fully in a later section) isolates rare library virions displaying domains that happen to bind a “selector”—a molecule, collection of molecules, or other substance chosen by the investigator. The ability of virions displaying a protein domain to replicate is essential to affinity selection, as explained in the next section.
The articles discussed in this review emphasize phage display's most conspicuous enterprise: phage antibody technology. Here, the displayed entity is an antibody domain such as a single-chain variable fragment (scFv) or an antigen-binding fragment (Fab), and the selector is a target antigen or mixture of antigens. Many of the articles featured in this review are from an accompanying collection, called Advances in Phage Display, edited by Gregg Silverman of New York University School of Medicine, Christoph Rader of the University of Florida, and Sachdev Sidhu of The Anvil Institute in Ontario. The collection is a successor to Phage Display: A Laboratory Manual, edited by Carlos Barbas and Dennis Burton of the Scripps Research Institute, Jamie Scott of Simon Fraser University, and Silverman, and published by the Cold Spring Harbor Laboratory Press in 2001 (Barbas et al. 2001). The entire Advances in Phage Display collection is available online at Cold Spring Harbor Protocols and can be accessed at https://cshprotocols.cshlp.org/.
This overview is divided into seven sections:
-
What Is a Phage Antibody?—a brief preview of phage antibodies
-
Filamentous Phage Structure and Function—essential information about the predominant phage species serving as phage display vectors
-
Phagemid Vectors for Phage Antibody Display—discussion of the predominant type of filamentous phage vector for display of antibody domains
-
Phage Antibody Libraries—review of design and construction of libraries with billions of phage clones, each displaying a different antibody domain with different binding specificity, and each represented by thousands, millions, or billions of genetically identical phages
-
Affinity Selection—explanation of exposing a solid surface coated with an antigen to a phage antibody library, in order to isolate very rare phages whose displayed antibody domains happen to bind the antigen with high affinity
-
Display of Peptides and Protein Domains Other than Antibodies—discussion of libraries displaying antigenic peptides, antibody-like binding domains, and other binding domains
-
Postscript: The C.A.R.L.O.S. Project—account of an emergency project using phage antibody display to develop chimeric antigen receptor T-cell (CAR T) therapy against a cancer afflicting Carlos Barbas, a key contributor to phage antibody technology from its earliest days
The articles in the collection serve not only individually as contributions to phage display methodology, but also collectively as a laboratory manual for students of an advanced phage display course that has been taught at the Cold Spring Harbor Laboratory since 1992, when phage display in its contemporary form, including phage antibody technology, was just emerging in a growing community of researchers. The course was first offered under the title “Phage Display of Combinatorial Antibody Libraries,” with Barbas and Burton as the founding instructors. It is now called “Antibody Engineering and Display Technologies,” with Silverman and Gianluca Veggiani of Louisiana State University as the current instructors. I refer to it here simply as the “Phage Display Course.”
In large measure, the Phage Display Course has been a joint project of the Cold Spring Harbor Laboratory's Courses Program and the Scripps Research Institute, which has been a center of phage antibody development from its earliest days and the source of key resources used in the course's laboratory investigations. Many of the instructors and lecturers, including authors represented in this collection, have worked at Scripps during their careers. These include in particular Barbas, Burton, Silverman (the longest serving instructor), and Rader (who, along with Barbas, assumed a major role in developing the pComb family of phagemids, leading vectors for phage antibody technology). I have tried not only to engage with the articles’ experimental data and logic, but also to touch on the broader intellectual culture that they are embedded in.
I assume that the reader is already familiar with the basic structure of antibodies and their interaction with antigens. Given only that background, I review the logic of the articles included in the collection, in the broader context of progress in phage antibody technology over the past 25 years.
WHAT IS A PHAGE ANTIBODY?
A phage antibody (Fig. 1) is an engineered phage in which an antibody domain—most often an scFv or Fab—is genetically fused to a phage protein that is exposed on the surface of the virion. The virion-borne antibody domain is thus accessible to antigens or other potential ligands in the surrounding medium. At the same time, the antibody domain is physically attached to a virion whose chromosome includes its coding sequence. The antibody domain is said to be “displayed” on the surface of the virion. Peptides and protein domains other than antibodies can be displayed on virions in the same way.
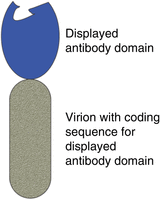
Schematic diagram of a phage antibody (not to scale). An antibody domain—most often a single-chain variable fragment (scFv) or an antigen-binding fragment (Fab)—is displayed on the surface of a virion (phage particle). The displayed antibody domain is exposed in the surrounding medium, where it can bind to an antigen. The virion's chromosome contains the coding sequence for the antibody domain it displays.
Physical linkage of the antibody domain to its coding sequence in the virion means that the domain can be propagated to prodigious numbers, simply by infecting host bacteria with virions displaying the domain. In effect, the antibody domain can replicate. That is what makes it feasible to isolate exceedingly rare phage clones whose displayed antibody domains happen to bind a chosen antigen from enormous phage antibody libraries comprising many billions of phage clones, displaying billions of different antibody domains. All the virions in the library are jumbled together in a single vessel. Rare antigen-binding clones are obtained, not via clone-by-clone screening, but rather via an affinity selection process, which is outlined in Figure 2. As explained in the legend, virions that have been affinity-selected must be propagated (“amplified”) to be useful.
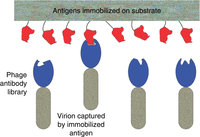
Isolation of rare antigen-binding antibody domains by affinity selection. An antigen of interest (red in the figure) is immobilized on a solid substrate of some kind. The antigen-coated substrate is exposed to the library—the “input” to the affinity selection process. Rare virions whose displayed antibody domains happen to bind the immobilized antigen are captured on the substrate surface, while all other virions—the overwhelming majority—remain free in solution. Virions that have not been captured are thoroughly washed away, leaving only the captured virions on the surface. The captured virions are released from the substrate in some manner, resulting in an “output” virion population that is greatly enriched for virions displaying antigen-binding antibody domains. No clone in the output population is present in sufficient numbers to permit meaningful characterization, or to serve as input to another round of capture and release. Instead, taking advantage of the physical link between the displayed antibody domain and its coding sequence in the phage genome, the output virions are greatly “amplified” (propagated) by infecting fresh bacterial host cells with output virions, and culturing the infected cells in growth medium. The amplified output population is then analyzed to identify clones of interest (those displaying antibody domains of interest), or used as input to another round of capture, release, and amplification.
FILAMENTOUS PHAGE STRUCTURE AND FUNCTION
The phage display concept has been realized with a number of phage platforms other than Ff filamentous phage, including λ, T4, T7, and Qβ (Istomina et al. 2024); each has key advantages. Nevertheless, the great majority of phage display applications still use the Ff family of filamentous phages (the very closely related natural strains M13, f1, and fd), and those phages have also been the platform for the Phage Display Course ever since its inception in 1992. This overview accordingly focuses on Ff filamentous phages.
The first chapter of the 2001 Phage Display: A Laboratory Manual was an authoritative review of filamentous phage structure and function by Robert Webster of Duke University (Webster 2001). Much of what we know about these phages was already clear then. Virions can easily be prepared at high purity. Electron microscopy (Fig. 3) reveals filamentous particles 6 nm wide and 900 nm long in the case of wild-type virions (the length of the virion can vary widely depending on the length of the DNA chromosome it contains). There are 11 phage proteins: five structural proteins that comprise the virion's cylindrical outer coat, and six proteins required for replication of the phage DNA and assembly of the virion. All but the tips of the cylindrical outer coat are formed by a fivefold helical array of thousands of copies of the 50-amino-acid major coat protein pVIII. The two tips of the virion are physically different: one pointy and one rounded, as depicted in high-resolution electron micrographs (Fig. 3). By 2001, it had been established that there are three to five subunits each of minor coat proteins pVII and pIX at the rounded tip, and three to five subunits each of minor coat proteins pIII and pVI at the pointy tip. All four minor coat proteins are now known to be present in exactly five copies each.
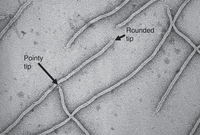
High-resolution negative-stained electron micrograph of filamentous phage virions. Putative pointy and rounded tips are indicated. Courtesy of Lee Makowski, Northeastern University, with permission.
Table 1 summarizes the domains of minor coat protein pIII, one of the minor coat proteins at the pointy tip, and a key component of phage display technology. An N-terminal domain of each pIII subunit is flexibly connected to the main body of the virion. A few of these domains can sometimes be discerned in electron micrographs, as exemplified by the four small, irregular bodies visible at the tip of the arrow pointing to the pointy tip in Figure 3. An X-ray crystallographic structure of the N-terminal domain has been determined (Lubkowski et al. 1999), revealing two subdomains, now called N1 and N2, attached to each other and to the C-terminal domain through flexible linkers. Domain N2 binds to the tip of a bacterium's F pilus to initiate infection; domain N1 subsequently interacts with the secondary receptor TolQRA in the bacterium's inner membrane, mediating entry of the phage DNA into the cytoplasm.
Domains of filamentous phage Ff minor coat protein pIII
Several features of minor coat protein pIII make it the most commonly used viral host protein for phage display, especially of antibody domains. Its flexible attachment to the main body of the virion ensures that peptides or proteins displayed on pIII are exposed in the surrounding medium and are thus able to react with antibodies, antigens, or other substances there. Like the other phage coat proteins, pIII is an inner membrane protein prior to being incorporated into the nascent virion. Its signal peptide, SS, targets it to the Sec secretion system (filamentous phages are secreted through the bacterial envelope without lysing the cell, as described below). The unfolded polypeptide is transported through the inner membrane until the first 21 residues of its 27-amino-acid transmembrane domain, TM, anchor it in the membrane, leaving only the last six amino acids exposed in the cytoplasm. Meanwhile, the 18-amino-acid signal peptide SS is cleaved from the polypeptide during secretion. The protein therefore folds into its functional conformation in the oxidizing environment of the periplasm between the inner and outer membranes. The periplasm is a putatively propitious environment for antibody domains displayed on pIII to likewise fold into their functional disulfide-stabilized conformations.
Jasna Rakonjac of Massey University and her colleagues have written a successor to Webster's review, reporting some stunning achievements since 2001 (see Overview: Structure, Biology, and Applications of Filamentous Bacteriophages [Rakonjac et al. 2024]). In particular, two advances have allowed the structures at the tips to be defined in detail (Conners et al. 2023). First, short, rigid “nanorods” have been constructed that have the same tip structures as wild-type virions. The viral DNA in these virions is much shorter than in wild-type virions, and the length of the particle is reduced in proportion. Instead of constituting ∼2.6% of a flexible wild-type virion, the tips constitute 28% of a rigid nanorod, making imaging much easier. Second, spectacular improvements in cryo-electron microscopy (cryoEM) allow detailed structures to be extracted computationally from great numbers of individually blurry images of nanorods in random orientations. These advances have allowed a detailed composite structure of the filamentous phage virion to be assembled (Fig. 4).
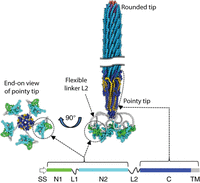
Composite structure of the Ff filamentous phage virion. Short segments of the virion near the rounded and pointy tips are depicted at the right (the wild-type virion is 150 times longer than its diameter). The circular single-stranded viral DNA, twisted into a long, thin helix, lies inside the protein coat; the length of the virion is variable, depending on the length of the DNA. The major coat protein pVIII subunits, thousands forming the bulk of the cylindrical outer coat, are bright blue; the minor coat protein pVII and pIX subunits (five each at the rounded tip) are salmon and purple, respectively; and minor coat protein VI subunits (five at the pointy tip) are gold. The domains of the minor coat protein pIII subunits (five at the pointy tip), which are mapped at the lower right and summarized in Table 1, are colored as follows: SS (absent from the virion) is white, N1 is green, flexible linkers L1 and L2 are gray, N2 is cyan, C is dark blue, and TM is gray. TM becomes part of domain C when incorporated into the virion, where it is colored dark blue like the rest of the domain. An end-on view of the pointy tip is shown at the left, with the five pIII subunits artificially arranged symmetrically around the central axis; in reality, these domains have highly variable orientations because of the flexibility of linker L2. Adapted from Overview: Structure, Biology, and Applications of Filamentous Bacteriophages [Rakonjac et al. 2024]) with permission from Cold Spring Harbor Laboratory Press.
Short peptides are typically displayed pentavalently (on all five pIII subunits), fused to the N terminus at the beginning of domain N1. As explained in the next section, in most phage antibodies, the antibody domain is displayed monovalently (on only one of the five pIII subunits); it can be fused either to the N terminus of N1 or to domain C through all or part of flexible linker L2, replacing domains N1, L1, and N2.
Progeny filamentous virions are not released into the medium by lysing the host cell, as in the majority of well-studied phage species. Instead, the circular single-stranded viral DNA, twisted into a long, thin helix, is secreted through the envelope, losing its intracellular coat of pV subunits and acquiring its extracellular coat of virion structural proteins from the inner membrane as it passes. Host bacteria survive and continue to divide, albeit at a reduced rate. The rounded tip of the virion, assembled at the beginning of the twisted DNA, emerges first through the envelope; the pointy tip, assembled at the end of the twisted DNA, emerges last. The length of the virion is determined by the length of the viral DNA, rather than by constraints imposed by the geometry of the coat. Domains N1, L1, and N2 of the five pIII subunits are connected to the pointy tip through their flexible L2 linkers, as depicted in Figure 4.
The channel through which the nascent virion is secreted is composed of phage proteins pIV, pI, and pXI. CryoEM has been deployed to determine the detailed structure of the outer membrane component of the channel, formed by a 15-member ring of pIV subunits (Conners et al. 2021; see also Overview: Structure, Biology, and Applications of Filamentous Bacteriophages [Rakonjac et al. 2024]). Figure 5 depicts a nascent progeny virion emerging, rounded tip first, through the channel. The near half of the ring of pIV subunits (colored green in Fig. 5) has been cut away, revealing the tight fit between the channel and the emerging virion. It is remarkable that the N-terminal domains of the pIII subunits at the pointy tip of the nascent virion are able to pass through this channel, especially when they display extra antibody domains. Despite this remarkable research, it is not yet possible to formulate clear rules governing limitations on foreign peptides or protein domains that can be successfully displayed on the virion surface.
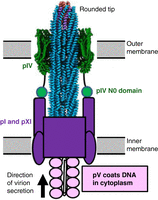
Schematic diagram of a nascent virion in the process of being secreted through the cell envelope. The outer membrane component of the channel, composed of a ring of 15 pIV subunits, is colored dark green. The front half of the outer membrane channel has been cut away in the image, in order to reveal the tight fit between the emerging virion and the interior of the channel. The inner membrane component of the channel, colored dark purple, is composed of pI and pXI subunits. A small N-terminal domain, N0, of each pIV subunit interacts with the inner-membrane component through a flexible linker (only two of the 15 N0 domains are depicted). As the nascent virion passes through the inner membrane, its DNA chromosome loses its cytoplasmic coat of phage pV subunits (light purple in the image) and acquires its sheath of coat proteins from the inner membrane. The chromosome is oriented with its packaging signal (hairpin [A] in Fig. 6) at the rounded tip. When the end of the chromosome is reached, five subunits each of minor coat proteins pIII and pVI are added to form the pointy tip of the virion. Adapted from Overview: Structure, Biology, and Applications of Filamentous Bacteriophages (Rakonjac et al. 2024) with permission from Cold Spring Harbor Laboratory Press.
Foundational investigation of Ff phage replication by Kensuke Horiuchi and Norton Zinder of Rockefeller University (Zinder and Horiuchi 1985) defined a 508-nt segment of the genome, called the intergenic region (IR), that orchestrates phage replication and assembly. It contains no coding sequence, but three essential cis-acting functions: a packaging signal required for efficient virion secretion, the minus-strand origin of replication, and the plus-strand origin of replication (Fig. 6). As explained in the next section, most phage antibody vectors are plasmids containing the IR (often referred to as the “origin”) and a recombinant pIII gene that includes the coding sequence for the antibody domain, but no other phage sequences.
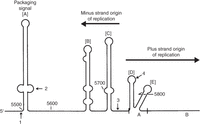
Intergenic region (IR) of phage f1 single-stranded viral DNA (the plus strand), comprising three cis-acting elements: packaging signal, minus-strand origin, and plus-strand origin. The plus-strand origin is divided into functional domains A and B (only part of domain B is included). [A]–[E] indicate the secondary structure hairpins in the plus strand. Four-digit numbers indicate nucleotide positions in the f1 genome. Arrows point to the end of the pIV coding sequence (1), the transcription terminator (2), the initiation site of the minus-strand RNA primer (3), and the origin of plus-strand synthesis (4). Modified from Zinder and Horiuchi (1985) with permission from the American Society for Microbiology.
PHAGEMID VECTORS FOR PHAGE ANTIBODY DISPLAY
Phagemid vectors such as pBluescript (Alting-Mees and Short 1989) are hybrids between ordinary recombinant DNA plasmids and filamentous phages. Like standard recombinant DNA plasmids, pBluescript has a plasmid origin of replication, a gene conferring resistance to an antibiotic (ampicillin), and a short multiple cloning site (MCS) with unique cleavage sites for several restriction enzymes. The MCS facilitates inserting a foreign DNA payload into the plasmid. The feature that defines pBluescript as a phagemid is inclusion of the filamentous phage IR (Fig. 6). The IR is ordinarily quiescent, but if the phagemid-bearing cell is infected by a fully functional filamentous phage (called the helper phage—helper for short), phage proteins act not only on the helper's own IR, but on the phagemid IR as well. Helpers usually include a gene conferring resistance to kanamycin; culturing the helper-infected cells in medium containing both ampicillin and kanamycin ensures that only cells bearing both genomes replicate. Two different single-stranded DNAs are produced and packaged into virions by the doubly resistant cells: the helper chromosome and the phagemid chromosome.
Phagemids provide an attractive platform for phage display (Qi et al. 2012), as exemplified by the pComb3 family of phagemids, introduced by Barbas and his colleagues (Barbas et al. 1991). They installed an expression cassette driven by the Escherichia coli lac promoter operator in a pBluescript-based backbone. The expressed coding sequence has cloning sites that allow a foreign protein or peptide, particularly an antibody domain, to be fused to pIII domain C, through all or part of pIII's flexible linker L2 (the domains of pIII are explained in Table 1 and Fig. 4).
There is an important advantage to fusing the antibody domain to pIII domain C, bypassing pIII's N-terminal domains N1 and N2. Expression of N2 suppresses synthesis of the F pilus, the primary receptor for filamentous phage infection; N1 suppresses synthesis of the secondary receptor TolQRA. This means that if the antibody–pIII fusion protein includes N1 and N2, its expression must be repressed before the host bacteria can be infected with helper phages. Adequate repression is difficult to achieve in the case of the lac promoter, especially at the optimal growth temperature of 37°C. Eliminating N1 and N2, as in the pComb3 family of vectors, avoids this problem. Nevertheless, expression of the fusion protein from the lac promoter is seldom derepressed by adding the inducer isopropyl β-d-1-thiogalactopyranoside (IPTG) to the culture medium. The usual reason given for thus relying on leaky expression is to reduce expression of the antibody domain so that it is displayed predominantly monovalently (on only one of the five pIII subunits). However, a more important reason is probably that even modest overexpression of the protein is highly toxic to the cell.
Most phage antibody libraries today, including those created in the Phage Display Course, are constructed in pComb3 family vectors, in conjunction with improved helper phages such as VCSM13 (Russel et al. 1986). The pComb3 family is reviewed by Christoph Rader, a leader in the development of phage antibody libraries, starting when he was a colleague of Barbas at Scripps (see Overview: The pComb3 Phagemid Family of Phage Display Vectors [Rader 2024b]).
When a pComb3-bearing cell is infected with a helper phage, both the wild-type pIII encoded by the helper and the recombinant pIII encoded by the phagemid are produced and incorporated into both helper and phagemid virions. In the recombinant protein, the cloned foreign protein or peptide replaces the normal N-terminal domains N1, L1, and N2. Even though the recombinant pIII is missing the domains that mediate the infection process, the virions are still infective because some of their pIII subunits are the wild-type protein encoded by the helper.
Antibody domains (either Fabs or scFvs) are almost always displayed monovalently (on only one pIII subunit) or not at all, for two reasons. First, wild-type pIII subunits encoded by the helper phage greatly outcompete recombinant pIII subunits encoded by the phagemid for incorporation into nascent virions. Second, only a minority of the phagemid-encoded pIII subunits that are incorporated into completed virions have intact antibody domains, because of the susceptibility of foreign proteins to degradation in the periplasm or cytosol.
Figure 7 diagrams an example of a coding sequence whose expression is driven by the lac promoter in a pComb3 family phagemid. In this construct, it is a chicken scFv that is fused to domain C of pIII. This format of the scFv–pIII genes in the phage antibody library is used by students in the Phage Display Course today.
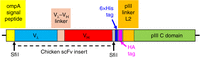
Schematic diagram of the coding sequence in a phage antibody construct in a pComb3 family phagemid vector (Andris-Widhopf et al. 2000). The coding sequence for a chicken single-chain variable fragment (scFv) is inserted between the SfiI cloning sites. The colored blocks together constitute the coding sequence for a recombinant protein in which a chicken scFv is fused to the C domain of pIII (including TM) through the last 27 amino acids of pIII's flexible linker, L2. The N-terminal domains N1, L1, and N2 are absent. The gray blocks are spacers with no specified function in the protein. The entire coding sequence spans 463 amino acids if Vλ and VH are the germline genes (the number can be slightly different if Vλ or VH have been modified by somatic gene conversion, as explained in the next section). The 21-amino-acid ompA signal peptide (yellow), like pIII's natural signal peptide, targets the protein to the Sec secretion system, and is cleaved off as the recombinant polypeptide passes through the inner membrane into the periplasmic space. The 6×His and HA tags allow virions to be specifically purified by immobilized metal affinity chromatography (IMAC) or affinity chromatography with an antibody against influenza hemagglutinin protein, respectively.
PHAGE ANTIBODY LIBRARIES
A phage antibody library is a mixture of millions or billions of phage clones, each clone displaying a different antibody domain, with different antigen-binding specificity. Each clone is represented by hundreds, thousands, or millions of genetically identical virions. The whole population is mixed together in a single vessel.
Antibody domains can be displayed on phages in different formats (Istomina et al. 2024):
-
Fab fragments are natural compact domains of antibodies, each including a light chain and the first two segments (VH and CH1) of a heavy chain (see Protocol: Cloning, Expression, and Purification of Phage Display-Selected Fab for Biophysical and Biological Studies [Cyr et al. 2024], Protocol: Generation of Antibody Libraries for Phage Display: Chimeric Rabbit/Human Fab Format [Peng and Rader 2024a], Protocol: Generation of Antibody Libraries for Phage Display: Human Fab Format [Peng and Rader 2024b], and Introduction: Generation and Selection of Phage Display Antibody Libraries in Fab Format [Rader 2024a]).
-
scFv constructs consist of only the VH and VL segments of an Fab, connected in either order by a flexible linker (they are illustrated in Fig. 7 and discussed in the first subsection below).
-
Diabodies consist of two scFv domains (identical or different) connected by a short linker.
-
Single-domain antibodies (sdAbs) consist of the heavy-chain variable region VH from a class of camelid antibodies that contain no light chain and no heavy-chain CH1 domain.
In almost all cases but sdAbs, heavy- and light-chain V region repertoires are created separately and joined in random combinations to make the final constructs, containing both heavy- and light-chain V regions. The number of possible combinations of heavy- and light-chain V regions is orders of magnitude greater than in the component V region repertoires.
The larger and more diverse the library, the more likely it is that a few of its displayed antibody domains will have high affinity for a target antigen of interest (called the selector). Such clones are isolated from the library by affinity selection (see Fig. 2), which is detailed in the next section. The three subsections of the current section review a few examples of phage antibody libraries, displaying antibody domains with various formats.
Phage Antibody Libraries from Immunized Humans, Chickens, and Other Animals
In many phage antibody projects, antibody domains are obtained from animals that are immune to one or more antigens of interest, either because they have been artificially immunized with a vaccine or other immunogen or because they have been naturally infected by a pathogen. For example, Jing Yi Lai and Theam Soon Lim of Universiti Sains Malaysia have reviewed projects in which the donors are humans who have been infected by viruses, bacteria, or parasites (Lai and Lim 2020).
Junho Chung of Seoul National University and his colleagues introduce the chicken immune system (see Introduction: Generation of Chicken Antibody Libraries and Selection of Antigen Binders [Yang et al. 2025c]) and detail the construction and use of a chicken phage antibody library displaying scFv domains against a chosen antigen (see Protocol: Chicken Immunization Followed by RNA Extraction and cDNA Synthesis for Antibody Library Preparation [Yang et al. 2025a], Protocol: Generation of a Phage Display Chicken Single-Chain Variable Fragment Library [Yang et al. 2025b], Protocol: Preparation of VCSM13 Helper Phage for Display Library Reamplification and Bio-Panning [Yang et al. 2025d], and Protocol: Selection of Antigen Binders from a Chicken Single-Chain Variable Fragment Library [Yang et al. 2025e]). The project that the students carry out in the Phage Display Course largely follows these articles, using a pComb3 family phagemid vector developed at Scripps (Andris-Widhopf et al. 2000; Cary et al. 2000).
The chicken antibody system differs from the human and mouse systems in several respects that make library generation simpler, and therefore particularly appropriate for the Phage Display Course (see Introduction: Generation of Chicken Antibody Libraries and Selection of Antigen Binders [Yang et al. 2025c]). There are only two immunoglobulin loci in chickens, one for the heavy (H) chain and one for the λ light chain; there is no κ light chain. There is only a single functional germline VH gene at the H locus, followed by four diversity (D) segments and a single joining (J) segment. Diversity is generated by VDJ joining and multiple somatic gene conversion events with more than 80 upstream VH pseudogenes. There is also only a single functional Vλ gene at the λ locus, followed by a single J segment. Diversity is generated by VJ joining and multiple somatic gene conversion events with 25 upstream Vλ pseudogenes. At both loci, there is no sequence variation near the end of assembled V genes in antibody-producing B cells, and very limited diversity near the beginning. The entire repertoire of VH genes can be amplified with only three forward primers combined with a single reverse primer, and the same is true for the Vλ gene repertoire (see Protocol: Generation of a Phage Display Chicken Single-Chain Variable Fragment Library [Yang et al. 2025b]). Most of the repertoire can be captured with a single forward primer for Vλ genes and a single forward primer for VH genes, as is done in the Phage Display Course. This simplicity is in sharp contrast to the profusion of primers required to capture the repertoire of VH, Vλ, and Vκ genes in human B cells (see Protocol: Generation of Antibody Libraries for Phage Display: Human Fab Format [Peng and Rader 2024b]). Constructing an scFv library from chickens is therefore relatively straightforward, making it a practical exercise in the Phage Display Course.
Chicken scFv libraries are constructed from laying hens that have been hyperimmunized with the antigen of interest (Andris-Widhopf et al. 2000; Cary et al. 2000; see also Protocol: Generation of a Phage Display Chicken Single-Chain Variable Fragment Library [Yang et al. 2025b]). The progress of the hens’ immune response to the antigen is assayed by titering immunoglobulin in blood or in egg yolks, each of which contains up to 100 mg of IgY (the chicken homolog of mammalian IgG). When the titer is adequate, the hens are euthanized, and RNA is extracted from lymphoid organs, such as spleen and bone marrow. The RNA is reverse-transcribed to create cDNA molecules that include VH and Vλ coding sequences. The cDNAs in turn serve as templates for PCR amplification of the hens’ entire VH and Vλ repertoires, using the simple primer sets described in the previous paragraph. The primers are designed to create a flexible linker between the VH and Vλ amino acid sequences so that they fold into a functional scFv domain, and to install suitable flanking restriction sites so that the scFv construct can be inserted into the phagemid vector in the correct reading frame, as illustrated for SfiI cloning sites in Figure 7. Vector DNAs bearing the scFv inserts are transfected into E. coli host cells; when the transfected cells are infected with helper phages, they release virions bearing scFv domains on their recombinant pIII subunits. These virions are the input to the first round of affinity selection.
Chicken phage antibody libraries can be smaller than the all-purpose libraries discussed in the next subsection. All-purpose libraries, either synthetic or representing large numbers of individual donors, are intended to include antibody domains that bind almost any chosen antigen of interest. Hundreds of millions to hundreds of billions of independent phage clones are required to meet this goal. A large portion of the library must be used to ensure that individual clones are represented by a sufficient number of virions. A chicken phage antibody library, in contrast, represents the antibody repertoire of chickens that have been hyperimmunized with the antigen of interest, so that a substantial number of their circulating antibodies are specific for the immunogen. A few million independent clones usually suffice to include high-affinity antibodies specific for the antigen.
More than 20 years ago, a few hens were hyperimmunized with the dye fluorescein conjugated to bovine serum albumin (FL-BSA), as described previously (Andris-Widhopf et al. 2000). The hens made not only antibodies that reacted with BSA, but also antibodies that reacted with the fluorescein dye. Small molecules like fluorescein that provoke an antibody response when conjugated to a protein antigen are called haptens; the proteins they are conjugated to are called carriers. RNA was extracted from the hens’ spleens and bone marrows and frozen in aliquots for long-term storage. Aliquots have served as the starting point for the Phage Display Course's student projects ever since. The availability of that RNA stock, as well as the small size of the library required to represent the repertoire of anti-fluorescein scFv domains, have made it practical for the students to complete their project in the 2 weeks available.
More generally, hyperimmunizing a few hens with a chosen antigen of interest, euthanizing them and harvesting their spleens and bone marrow, and constructing a small phage antibody library and affinity-selecting antibodies against the antigen or hapten using easily obtained supplies and standard protocols—that can be an attractive alternative to constructing a large all-purpose library (next subsection), if such a resource is unavailable or too costly to buy or construct.
All-Purpose Phage Antibody Libraries: Naive and Synthetic
All-purpose libraries are designed to represent such a diversity of independent clones that they are likely to include high-affinity antibody domains specific for almost any antigen of interest. There are two major types (Almagro et al. 2019): natural naive libraries, meant to express the entire natural antibody repertoire of many (sometimes as many as 200) donors (human or nonhuman) who have not been purposely immunized with an antigen of interest, and synthetic libraries, in which randomized synthetic complementarity-determining regions (CDRs) are installed in the backbone of an animal's (usually a human's) natural antibody variable regions.
A 2017 review summarizes anti-pathogen antibodies that have been affinity-selected from dozens of natural naive libraries assembled from RNA pooled from many healthy human donors (Chan et al. 2017). A series of articles by Haiyong Peng (University of Florida) and Rader details construction of naive human libraries in Fab format (see Protocol: Generation of Antibody Libraries for Phage Display: Chimeric Rabbit/Human Fab Format [Peng and Rader 2024a], Protocol: Generation of Antibody Libraries for Phage Display: Human Fab Format [Peng and Rader 2024b], Protocol: Generation of Antibody Libraries for Phage Display: Library Reamplification [Peng and Rader 2024c], Protocol: Generation of Antibody Libraries for Phage Display: Preparation of Electrocompetent E. coli [Peng and Rader 2024d], Protocol: Generation of Antibody Libraries for Phage Display: Preparation of Helper Phage [Peng and Rader 2024e], Protocol: Phage Display Selection of Antibody Libraries: Panning Procedures [Peng and Rader 2024f], and Protocol: Phage Display Selection of Antibody Libraries: Screening of Selected Binders [Peng and Rader 2024g]).
Like natural naive libraries, synthetic libraries do not entail immunizing live animals or humans. However, they also have a critical advantage: Antibodies against self-antigens are not censored by immunological tolerance. Tolerance might block discovery of high-affinity human antibody domains against human antigens, for instance. Human anti-human antibodies are an increasingly important class of pharmaceuticals.
Three articles by Sidhu, Veggiani, Maryna Gorelik (University of Waterloo), and Shane Miersch (University of Waterloo) review the “minimalist” approach to synthetic human libraries that the Sidhu laboratory has been pursuing for more than two decades (see Overview: Structural Survey of Antigen Recognition by Synthetic Human Antibodies [Gorelik et al. 2025], Introduction: Beyond Natural Immune Repertoires: Synthetic Antibodies [Veggiani and Sidhu 2024a], and Protocol: Generation and Selection of Synthetic Human Antibody Libraries via Phage Display [Veggiani and Sidhu 2024b]). The overall goal is to develop a practical way to construct libraries from which promising therapeutic antibodies for any chosen antigen can be affinity-selected. They focus on two desirable physical properties of such antibodies: high affinity for the antigen and high “developability,” meaning possession of properties such as solubility and stability that facilitate manufacture and favorable pharmacokinetic behavior. The defining feature of the minimalist approach is using an already existing therapeutic antibody with favorable developability as the sole scaffold for mutagenesis, thus greatly simplifying library construction and hopefully increasing the chance that antibody domains selected from the library will share the favorable developability of the chosen scaffold antibody. The scaffold for many minimalist projects has been the anti-HER2 antibody trastuzumab. Minimalism's key assumption is that high affinity can be achieved without exploiting the entire natural diversity of scaffolds in natural antibodies. This assumption has been largely vindicated in the sequel.
Minimalist libraries are constructed by introducing random mutations in strategically chosen codons in the scaffold antibody's six CDRs. One of the three articles mentioned above provides a general protocol for constructing such a library (see Protocol: Generation and Selection of Synthetic Human Antibody Libraries via Phage Display [Veggiani and Sidhu 2024b]):
-
A “stop template” is cloned in a phagemid vector. The stop template encodes the scaffold sequence (e.g., trastuzumab), but with a stop codon at each position chosen for mutagenesis.
-
The resulting vector is transfected into a mutant E. coli host that incorporates deoxy-U (dU) in place of many T nucleotides. The transfected cells are infected with helper phages and incubated in medium that selects for both phagemid and helper. Virions are prepared from the culture supernatant (there are no phagemid-encoded pIII molecules on these virions).
-
Single-stranded plus-strand DNA is prepared from the virions; that DNA has numerous dU nucleotides in place of T nucleotides.
-
Synthetic minus-strand mutagenic oligonucleotides, one for each of the six CDRs, are hybridized to the plus-strand virion DNA. These oligonucleotides have randomized sense codons opposite each of the stop codons in the plus strand. The minus strand is then closed by incubation with DNA polymerase and ligase. This reaction creates double-stranded heteroduplex DNA whose plus strand has numerous dU nucleotides and stop codons at the sites chosen for mutagenesis, and whose minus strand has no dU nucleotides and has randomized sense codons at the sites chosen for mutagenesis. Many (but not all) of the heteroduplexes will be covalently closed circles. This DNA is stored frozen in aliquots for use in preparing fresh libraries, as described in the next bullet.
-
To prepare a new library, an aliquot of the heteroduplex DNA is electroporated into electrocompetent E. coli prepared from helper-infected cells. The electroporated cells are cultured in medium that selects for both helper and phagemid genomes. The cells do not incorporate dU nucleotides into DNA, and have an intact dU repair system that cuts the plus strand into small segments. The great majority of replicating DNAs, and of the DNAs in the secreted virions, are therefore derived from covalently closed circular minus strands, in which the stop codons have been replaced with randomized sense codons. Phages displaying antibody domains that bind a chosen antigen can then be affinity-selected from the resulting library.
By now, many human antibodies that have been affinity-selected from minimalist libraries are available for comparison to natural antibodies (see Overview: Structural Survey of Antigen Recognition by Synthetic Human Antibodies [Gorelik et al. 2025]). These authors compared 50 antibodies affinity-selected from minimalist libraries with 50 natural antibodies from the same VH and Vκ subgroups. High-resolution structures of the antibody–antigen interaction were available for all 100 antibodies. By several broad criteria, the structures of the CDRs in the two types of antibody and the interaction of the CDRs with the antigen were strikingly similar. Figure 8 shows an example of the authors’ analysis. All CDR amino acids defined as “core” in the figure are represented in at least 60% of the structures analyzed. To facilitate graphical comparison, those core amino acids are mapped onto the backbone of the trastuzumab Fab (even though the backbone conformation differs significantly from one antibody to another). The size of each colored circle in the figure is proportional to the percent of structures in which that core amino acid participates significantly in the CDR–antigen interface; amino acids for which this percent was <20 are omitted. The striking similarity of the graphs for synthetic and natural antibodies implies that synthetic and natural CDRs interact with antigen in very similar ways. The authors argue that their results suggest considerable commonality of CDR structure, regardless of the backbone in which they reside, and that high-affinity antibodies can be selected from libraries in which only one of the natural antibodies’ myriad scaffolds is represented.
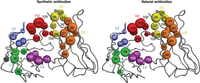
Graphical representation of the interaction between “core” complementarity-determining region (CDR) amino acids and antigen in synthetic and natural antibodies. The definition of core amino acids and their representation in the figure are explained in the text. Figure reprinted from Overview: Structural Survey of Antigen Recognition by Synthetic Human Antibodies (Gorelik et al. 2025) with permission from Cold Spring Harbor Laboratory Press.
A series of four articles by Mary Ann Pohl of Ailux Biologics and Juan Almagro of GlobalBio, Inc., describe an elaboration of the minimalist concept that they call semisynthetic libraries (see Protocol: Semisynthetic Phage Display Library Construction: Design and Synthesis of Diversified Single-Chain Variable Fragments and Generation of Primary Libraries [Almagro and Pohl 2024a], Protocol: Semisynthetic Phage Display Library Construction: Generation of Filtered Libraries [Almagro and Pohl 2024b], Protocol: Semisynthetic Phage Display Library Construction: Generation of Single-Chain Variable Fragment Secondary Libraries [Almagro and Pohl 2024c], and Introduction: Considerations for Using Phage Display Technology in Therapeutic Antibody Drug Discovery [Pohl and Almagro 2024]).
Such libraries are created in three stages:
-
Construction of four primary libraries: Each of the four primary scFv libraries is constructed on one of four human Vκ scaffolds in the N-terminal position, connected by a flexible linker to a common human VH scaffold in the C-terminal position. The VH scaffold's V gene segment is followed by a mixture of 90 combinations of human diversity (D) and joining (J) gene segments. The sequence from the beginning of CDR3 to the end of FR4 (corresponding to the last codon of the V gene segment through the D and J gene segments) is a temporary placeholder that will be replaced in construction of the secondary libraries (last bullet). Hopefully, the very limited variation in the placeholder's sequence and length is sufficient for the purposes of “filtration” (next bullet). Meanwhile, random variation is introduced at strategically chosen codons in all three CDRs of the Vκ scaffolds and CDR1 and CDR2 of the VH scaffold. The four primary libraries are assembled by chemical DNA synthesis and stored as frozen aliquots.
-
Construction of four filtered libraries: The “filtered” libraries consist of members of the primary libraries whose scFv domains are resistant to heat shock and are therefore presumably well folded into a stable native three-dimensional structure. Construction begins by PCR-amplifying an aliquot of each of the four primary libraries, splicing the amplified DNA into the SfiI cloning sites of a pComb3-like phagemid vector, and propagating the library by electroporating the spliced DNA into an E. coli host and infecting with a helper phage. Partially purified phagemid virions are prepared from the four culture supernatants. The virions are then heat-shocked and cooled to allow renaturation of the displayed scFvs (the virions themselves are impervious to the heat shock). Superparamagnetic beads coated with protein L are used to affinity-purify virions from each of the four heat-shocked virion preparations, discarding virions that are not captured on the beads. Protein L binds FR1 of all four Vκ scaffolds in well-folded scFvs. The affinity-purified virions are therefore enriched for virions displaying particularly stable scFvs. Virions from each of the four libraries are released from the beads and propagated separately by infecting E. coli host cells. Aliquots of phagemid-bearing cells are frozen to serve as input to construction of the secondary libraries (next bullet).
-
Construction of four secondary libraries: An aliquot of frozen phagemid-bearing cells from each of the four filtered libraries is thawed and cultured in medium that selects for the phagemid (the cells are not infected with helper phages, and the medium does not select for helpers). Phagemid DNA is extracted from the four cultures and partially purified. A secondary library is prepared from each of the four phagemid DNAs by replacing the 90 placeholder CDR3-FR4 sequences at the end of the VH regions with a mixture of hundreds of millions of natural CDR3-FR4 sequences prepared from 200 adult human donors. Replacement is mediated by a highly conserved nucleotide sequence at the end of human VH FR3. The conserved sequence serves as a PCR priming site for fusing the natural and filtered library sequences. Each of the four fused DNAs is spliced into the SfiI cloning sites of the pComb3-like phagemid vector, which is electroporated into E. coli host cells. The phagemid-bearing cells are infected with helper phages and cultured. Phagemid virions prepared from the culture supernatants comprise the four secondary libraries.
The resulting ensemble of four semisynthetic libraries is called “ALTHEA Gold Plus Library.” It is a successor to the “ALTHEA Gold Library,” an ensemble of two semisynthetic libraries, representing two, rather than four, human Vκ scaffolds (Valadon et al. 2019); for the ALTHEA Gold Library, filtration was accomplished with protein A rather than protein L.
High-affinity, conformationally stable scFvs have been affinity-selected from both the ALTHEA Gold Library (Valadon et al. 2019; Pedraza-Escalona et al. 2021; Dao et al. 2022a,b) and the ALTHEA Gold Plus Library (Guzmán-Bringas et al. 2023; Mata-Cruz et al. 2025). Mata-Cruz and her colleagues, for example, sought human scFvs that bind human CD36, a multifunctional membrane protein that is implicated in numerous diseases and is the target of drug development projects (Guerrero-Rodríguez et al. 2022). After three rounds of affinity selection using the extracellular domain of human CD36 as selector, they analyzed phagemids from 90 randomly selected clones. Fifty-seven of the phagemids, representing four distinct scFvs, bound strongly and specifically to the CD36 selector. All but 17 of the 90 phagemids, including all 57 that bound strongly to the selector, bound strongly to protein L, indicating that their displayed scFvs were predominantly in a natural immunoglobulin conformation. One of the four distinct scFvs, called D11, was shown to mimic the anti-metastatic effects of a mouse monoclonal antibody that binds both mouse and human CD36 (Pascual et al. 2017). These results commend D11 as a possible pharmaceutical lead. Whether or not that promise is realized, the results demonstrate the effectiveness of the Althea Gold Plus Library as a source of well-folded, high-affinity scFvs for a chosen selector.
Affinity Maturation
In the course of a natural antibody response, the affinity of the antibodies for the antigen increases progressively as a result of somatic mutation of the V regions in the responding B cells, along with clonal competition and increasingly stringent selection for high affinity by the antigen. An artificial analog of natural affinity maturation can be imposed on phage antibody clones that emerge from affinity selection with the antigen of interest (next section). DNA sequences from the selected clones are mutagenized at strategic positions and used to create a library of mutagenized phages. The mutagenized library is subjected to increasingly stringent affinity selection, in the hope of creating phage antibody clones with even higher affinity (Tiller et al. 2017).
AFFINITY SELECTION
In the phage antibody context, as diagrammed in Figure 2, affinity selection is accomplished by immobilizing the antigen of interest on a substrate of some kind, and then reacting the antigen-coated substrate with a library of phage antibodies (see Overview: Principles of Affinity Selection [Smith 2024]). Virions displaying antibody domains that happen to bind the immobilized antigen are captured on the substrate surface, while other virions—the overwhelming majority—remain free in solution. Virions that have not been captured are washed away, and the captured virions are released from the surface; the released virions are the unamplified output of affinity selection. The output virions are then used to infect fresh bacterial cells, which are cultured in growth medium; the resulting progeny virions constitute the amplified output. Each virion in the unamplified output is represented by millions or billions of genetically identical progeny virions in the amplified output. The amplified output serves as the input virions for another round of affinity selection or is analyzed to identify promising clones for further investigation.
A generic vocabulary is introduced here in order to summarize the principles of affinity selection in general, not just in the particular case of affinity selection from phage antibody libraries. The immobilized species (an antigen in the case of phage antibody projects) is referred to generically as the “selector.” The species that is displayed on the surface of a virion, and potentially binds the immobilized selector (an antibody domain in the case of phage antibody projects), is referred to generically as a “peptide” regardless of size; whether the peptide is a protein, a protein domain, or a small peptide is to be understood in context. Using this vocabulary, a few general principles of affinity selection were explained (see Overview: Principles of Affinity Selection [Smith 2024]) and are discussed here.
Yield Versus Stringency
Theoretically, the yield of a phage clone from a round of affinity selection is the ratio of the number of virions of that clone in the input to that round to the number of virions of that clone in the unamplified output of that round. It is not generally possible to measure yield directly, but it is possible to estimate the relative yields of different phage clones bioinformatically, as explained in the final subsection of this section.
There are two types of yield:
-
Specific yield is dependent on the identity of the displayed peptide. The obvious reason for high specific yield is that the clone in question displays a peptide with high affinity for the selector—exactly the desired property in an affinity selection project. Such specific clones are selector-specific, as are their yields. However, some clones owe their relatively high specific yields to something other than high affinity of their displayed peptides for the selector. One example is a clone whose displayed peptide binds the substrate, rather than the selector. Such clones are examples of selector-unrelated phages, or SUPs, which are the subject of the next subsection.
-
Nonspecific background yield is independent of the identity of the displayed peptide. Some virions, whatever peptide they display, end up in the output just by chance, because no physical separation process is perfect. Nonspecific background yield is very low in affinity selection, but it is not zero.
Stringency is the degree to which a round of affinity selection favors phage clones whose displayed peptides bind the immobilized selector with high affinity over clones whose displayed peptides bind the immobilized selector with low affinity.
High yield and high stringency are both desirable characteristics of a round of affinity selection, but they are mutually antagonistic: High stringency almost always entails low yield and vice versa. High yield is mainly promoted by a high surface density of selectors on the substrate, while high stringency is promoted by a low surface density of selectors on the substrate. In the Phage Display Course, for example, stringency of selection for scFvs that bind the fluorescein (FL) hapten selector could be adjusted by immobilizing mixtures of FL-conjugated ovalbumin (FL-Ova) and unconjugated Ova in different proportions on the substrate surface. The smaller the proportion of FL-Ova, the higher the stringency.
High yield is imperative in the first round of affinity selection, when the input is a large library with billions of clones, even though that entails low stringency. Each individual clone in such a library is represented by a limited number of genetically identical virions—10 or 100, say. That includes the most desired clones: those displaying peptides that bind the selector with high affinity. If stringency is so high that not even a single virion from such a clone is represented in the unamplified output of the first round of affinity selection, that clone cannot magically reappear in subsequent rounds.
Stringency can be greatly increased in the second and subsequent rounds. That is because even when yield in the first round is as high as practical (i.e., stringency is as low as practical), the overall yield is still quite low. The number of clones represented in the output is almost never more than 0.01% of the number of clones represented in the starting library, and usually is much lower. That means that each clone can be represented by at least 10,000 times more identical virions in the input to the second round than in the input to the first round, assuming that the total number of virions in the inputs to the first and second rounds are the same.
Selector-Unrelated Phages, or SUPs
SUPs are unwanted phage clones that increase in prevalence with successive rounds of affinity selection for reasons other than affinity of their displayed peptides for the selector (Thomas et al. 2010). They can be classified into two types.
-
Some SUPs reflect high specific yield during the capture or release stages of affinity selection (the capture, release, and amplification stages of affinity selection are described in the first paragraph of this section). For instance, the displayed peptide might bind some component of the affinity selection apparatus other than the immobilized selector—the substrate itself, for example.
-
Other SUPs reflect overreplication during the amplification stage in each round of affinity selection. This unwanted overreplication is specific for the displayed peptide, but it is seldom caused by the displayed peptide. Much more often, it is the result of a property that is determined by genetic alterations that are linked to the coding sequence for the displayed peptide in the same phagemid chromosome.
I argue that with a few exceptions, measures to eliminate SUPs from a library have limited success (see Overview: Principles of Affinity Selection [Smith 2024]). A more promising general approach today is to identify SUPs bioinformatically, with the aid of high-throughput sequencing (HTS) of the clones that emerge from an affinity selection project. HTS is already increasingly favored as the “primary readout” in affinity selection projects; that is, the first stage in analysis of the selected clones. The role of HTS in identifying phage clones of interest, while eliminating SUPs from consideration, is the subject of the next subsection. That subsection discusses HTS in the context of affinity selection from phage antibody libraries, but its application to other types of libraries and selectors should be apparent.
High-Throughput Sequencing (HTS) as Primary Readout in Affinity Selection Projects
A series of articles by Brandon DeKosky and his colleagues at the Massachusetts Institute of Technology introduces HTS (see Introduction: Beyond Single Clones: High-Throughput Sequencing in Antibody Discovery [Fahad et al. 2025b]) and details its use in antibody engineering (see Protocol: Antibody Data Analysis from Diverse Immune Libraries [Fahad et al. 2025a], Protocol: Clonal Lineage and Gene Diversity Analysis of Paired Antibody Heavy and Light Chains [Fahad et al. 2025c], and Protocol: Clonal Variant Analysis of Antibody Engineering Libraries [Fahad et al. 2025d]).
HTS can deliver partial sequences of the antibody domains displayed by millions of virions in the output of a round of affinity selection. That is usually enough for a thorough census of the selected antibody domains in the outputs of the first and second rounds of affinity selection, giving the number of times each clone occurs; that number is referred to as the clone's reads. A clone's reads divided by the sum of the reads for all clones in the output is a measure of the clone's prevalence. The amplified output of the first round of affinity selection is the input to the second round. Therefore, the ratio of a clone's prevalence in the amplified output of the second round of affinity selection to its prevalence in the amplified output of the first round is a measure of the clone's yield in the second round of affinity selection relative to the yields of the other clones in that round. If, following the advice in the first subsection above, stringency is greatly increased in the second round, clones whose displayed antibody domains bind the selector with high affinity are strongly favored over clones displaying low-affinity antibody domains. This profile of the clones’ relative yields in the second round may be enough to choose the most promising clones for further investigation. Promising clones can be recovered from the second-round output by PCR with clone-specific primers.
Not all clones with high second-round yields necessarily display antibody domains with high affinity for the selector. Some of them might be SUPs, as explained in the previous subsection. Unwanted SUPs can be distinguished from desirable high-affinity clones if affinity selections with multiple selectors are carried out in parallel from a common library. The parallel selections should be as identical as possible, apart from the identity of the selectors. SUPs reveal themselves because their yield is high in all (or almost all) the parallel selections, regardless of the selector.
DISPLAY OF PEPTIDES AND PROTEIN DOMAINS OTHER THAN ANTIBODIES
Although phage antibodies are the most conspicuous arena of phage display technology and the focus of the Phage Display Course's laboratory experiments, many other types of peptides and proteins have been displayed on filamentous phages, a few of which are described below.
Epitope Discovery
“Epitope discovery”—identification of peptides or protein domains that are recognized by antibodies—has been an important application of phage display technology from its earliest days (Cortese et al. 1994). Antibodies—either monoclonal or polyclonal—are used to affinity-select antibody-binding peptides from two types of phage display library: random peptide libraries (RPLs) and natural peptide libraries (NPLs). RPLs display synthetic peptides with randomized sequences; NPLs display the entirety or fragments of natural proteins.
Two articles by Gregg Silverman (see Introduction: Insights from the Study of B-Cell Epitopes of a Microbial Pathogen by Phage Display [Silverman 2025a] and Protocol: Cloning and Selection from Antigen Fragment Libraries for Epitope Identification [Silverman 2025b]), summarizing recent work by his group at New York University (Hernandez et al. 2020a,b), exemplify epitope discovery from NPLs. His laboratory has developed a suite of techniques for rapid identification of short peptide epitopes, for potential incorporation into modular vaccines against infectious diseases. To this end, genes for target pathogen proteins are randomly fragmented into small DNA pieces, which are cloned into a pComb-derived vector called pComb-Opti8. This vector displays the cloned peptides on subunits of the major coat protein pVIII through a flexible linker, rather than on subunits of minor coat protein pIII (as illustrated in Fig. 4). The resulting NPLs include DNA inserts in all possible reading frames in both orientations, but there is sufficient redundancy for many thousands of in-frame inserts, spanning the entire protein-coding sequence, to be represented. Monoclonal or polyclonal antibodies of interest are then used to affinity-select phage clones displaying protein fragments that bind those antibodies. Individual affinity-selected clones can be sequenced to identify the selected epitope-containing peptides. Alternatively, and increasingly, outputs of affinity selection are analyzed en masse by HTS, as outlined in the final subsection of the previous section.
The investigators’ “proof-of-principle” project targets the leucocidins, which are major virulence factors in invasive infection by Staphylococcus aureus, including by methicillin-resistant S. aureus (MRSA). The leucocidins, reviewed by Spaan et al. (2017), are eight-member β-barrel rings of alternating type S and type F subunits. Such rings kill cells in infected hosts by creating pores in the cell membrane, thus breaching the barrier between inside and outside. There are multiple leucocidins, with different type S and type F pairs. In most cases, the two types of subunits are secreted separately. The type S subunit binds a receptor on a target cell; then, additional type S subunits and type F subunits assemble into the β-barrel pore at the receptor site. Different leucocidins target different cell types, depending on the receptors they bind. Receptors have been identified for the major leucocidins, and X-ray crystallographic structures are available for many leucocidins and individual type S and type F subunits.
Two illustrative articles from Silverman's group report construction of gene fragment libraries from two type S subunits in the pComb-Opti8 vector, and affinity selection of phage-borne peptides from each library, using as selectors a mouse monoclonal antibody elicited by immunization with the respective unfragmented parent type S subunit (Hernandez et al. 2020a,b). Clone-by-clone sequencing (Hernandez et al. 2020b) or HTS analysis (Hernandez et al. 2020a) of the affinity-selected clones from each library identified a minimal epitope recognized by the selecting monoclonal antibody. Both epitopes were also recognized by antibodies from patients recovering from invasive S. aureus infection. The availability of the X-ray crystallographic structure of the type S subunits allows the minimal epitopes to be located in the native folded proteins. One epitope corresponds to a solvent-exposed β-turn at the tip of a β-hairpin (Hernandez et al. 2020a); the other corresponds to an entire solvent-exposed β-hairpin (Hernandez et al. 2020b).
Remarkably, when mice were immunized with these small peptides conjugated to keyhole limpet hemocyanin, they produced antibodies that bind not only the immunizing peptides, but also the intact parent proteins from which the peptides derive. This ability to elicit antibodies that react with the target pathogen protein in its native state has been called immunogenic fitness (Matthews et al. 2002); it is presumably a necessary condition for almost any effective vaccine component. It is not clear how general strong immunogenic fitness will be for short peptide epitopes affinity-selected from fragment libraries. For some parent proteins, many or most epitopes may be discontinuous, composed of amino acids from distant parts of the linear amino acid sequence. In any case, the logic of these investigators’ strategy does not depend on the size distribution of the protein fragments. A gene could be randomly fragmented into much larger pieces, some of which happen to encode peptides that fold into natural subdomains that are present on the intact protein. Discontinuous epitopes on such subdomains are likely to be shared by the pathogen or pathogenic protein itself, and therefore have good prospects of strong immunogenic fitness.
Display of Nonantibody Antigen-Binding Domains
Antibody V regions are folded β-sheets supporting three loops with diverse amino acid sequences at one end. The β-sheet is conformationally stable, serving as a secure “scaffold” that keeps the loops in place despite their diversity. It is the diversity of the loops that accounts for the antibodies’ ability to bind specifically to an enormous diversity of antigens. The loops that mediate binding by an antibody are called complementarity-determining regions (CDRs), because it is their sequence diversity that confers an almost unlimited diversity of complementarity (i.e., antigen-binding specificity) on antibodies.
Several stable nonantibody scaffolds have been explored for construction of “phage nonantibody libraries” (as I call them here) representing a great diversity of binding sites. From such libraries, specific, high-affinity ligands for a great diversity of selectors can be affinity-selected. I call the selectors “antigens” here to highlight the parallelism between nonantibody binding domains and canonical antibodies. Such nonantibody antigen-binding domains, like antibody V regions, are composed of a structurally stable scaffold holding in place CDRs with highly diverse sequences and antigen-binding specificities.
Akiko Koide and Shohei Koide of New York University Grossman School of Medicine summarize the construction and use of monobodies, whose scaffolds are fibronectin type III (FN3) domains (see Overview: Use of Phage Display and Other Molecular Display Methods for the Development of Monobodies [Koide and Koide 2024]). The FN3 motif is widespread in nature: Humans produce 4104 of them in 673 proteins, a few of which have served as scaffolds for monobodies. Like antibody V regions, FN3 domains are folded β-sheets, but with seven rather than nine β-strands. As in the case of antibody V regions, there are three loops at one end of the folded sheet, connecting pairs of anti-parallel β-strands. The amino acid sequences in these loops can vary greatly without weakening the folded β-sheet structure; that sequence diversity gives monobodies the ability to bind specifically to a great diversity of antigens. Unlike an antibody V region, a monobody is not paired with another monobody in a stable superstructure comparable to a Fab. They are expressed as stand-alone antigen-binding domains, like the VHH domains in some camelid antibodies (Muyldermans 2013).
The investigators set out to display monobodies fused to the C-terminal domain of filamentous phage pIII protein, using a phagemid vector in the pComb3 family (see “Phagemid Vectors for Phage Antibody Display”), and have explored other display formats as well. Efficient display required changing pComb3's ompA signal peptide to a signal peptide dependent on the signal recognition particle (SRP), so that the protein was secreted through the inner membrane cotranslationally, rather than post-translationally. They argue that this change was necessitated by one of the FN3 domain's chief attractions: its extraordinary noncovalent stability, which is not reinforced by an intradomain disulfide bond. Rapid formation of the folded structure in the cytoplasm would arguably interfere with post-translational secretion.
Stefan Ståhl and his colleagues at KTH Royal Institute of Technology in Stockholm have authored articles introducing affibodies, another type of nonantibody antigen-binding domain (see Protocol: Selection of Affibody Molecules Using Escherichia coli Display [Dahlsson Leitao et al. 2024], Protocol: Selection of Affibody Molecules Using Phage Display [Hjelm et al. 2024], Protocol: Selection of Affibody Molecules Using Staphylococcal Display [Löfblom et al. 2024], Protocol: Cloning of Affibody Libraries for Display Methods [Ståhl et al. 2024a], and Introduction: Engineering of Affibody Molecules [Ståhl et al. 2024b]). Here, the scaffold is a three-helix bundle, and the CDRs are not loops protruding from one end of the bundle, but rather 13 amino acids in helices 1 and 2 that protrude from one side of the bundle and are not required for its structural integrity. The investigators explain how to display affibodies on a pComb3 family phagemid vector, on the surface of E. coli bacteria, and on the surface of Staphylococcus carnosus bacteria. With only 58 amino acids, affibodies are smaller than any other antigen-binding domain, yet they can have subnanomolar and even subpicomolar affinities for their target antigens. These features commend them for biomedical applications requiring rapid and long-lasting binding to antigens in human subjects, along with rapid clearance of unbound molecules. For example, affibody ABY-25 has 76 pm affinity for the breast cancer marker HER2 (Feldwisch et al. 2010). When conjugated to the chelator DOTA and labeled with the positron-emitting radionuclide 68Ga, whose half-life is only 68 min, it allows metastatic lesions to be imaged at high resolution in HER2-positive breast cancer patients by positron emission tomography (Velikyan et al. 2019; Alhuseinalkhudhur et al. 2020).
Display of Specialized Protein Domains
Gregory Martyn of the University of Toronto and Veggiani describe libraries of modified Src homology 2 (SH2) domains, which bind phosphorylated tyrosine (pTyr) residues on specific receptor tyrosine kinases or other proteins (see Overview: Phage-Displayed SH2 Domain Libraries: From Ultrasensitive Tyrosine Phosphoproteome Probes to Translational Research [Martyn and Veggiani 2024]). The human genome includes 122 of these ∼100-amino-acid domains, present on intracellular signal transduction proteins. When an SH2 domain binds pTyr, the protein that it is part of is triggered to set in train a signal transduction cascade. Being able to exploit this specificity to detect or modify the behavior of specific regulatory pTyr residues could have many potential biomedical applications. However, although SH2 domains generally bind specifically to their cognate pTyr residues, their affinity is generally too weak for most applications. The article describes procedures for randomizing strategic codons in an SH2-coding sequence, using phage display to affinity-select variants with higher affinity or altered specificity.
An article by Chen T. Liang, Olivia Roscow, and Wei Zhang summarizes affinity selection of high-affinity ligands for chosen selectors from libraries of ubiquitin (Ub) variants (see Overview: Generation and Characterization of Engineered Ubiquitin Variants to Modulate the Ubiquitin Signaling Cascade [Liang et al. 2024]). Ub is a highly conserved 76-amino-acid protein present in virtually all eukaryotic cells. The article emphasizes how detailed knowledge of the structure and properties of Ub can guide affinity selection projects. Ub might well have been included in the previous subsection, as yet another attractive scaffold for “phage nonantibody libraries,” as I call them there. Following the pioneering work of the Sidhu laboratory (Ernst et al. 2013), however, almost all proteins chosen as selectors for Ub variants already interact specifically with Ub (Tang et al. 2023).
Ub is central to a sprawling network of cellular functions.
-
Ubiquitination—enzymatic covalent conjugation of Ub to target proteins, which are called “substrates”—is at the heart of the Ub system. It is carried out by an enzyme cascade composed of three types of enzymes acting in succession: E1 activating enzymes, followed by E2 conjugating enzymes, followed by E3 ligating enzymes. The human proteome includes two E1s (Barghout and Schimmer 2021), about 40 E2s (Stewart et al. 2016), and well over 600 E3s (Yang et al. 2021). It is the E3 ligating enzymes that are responsible for conjugating Ub to specific substrate proteins, creating conjugates with single Ubs, or branched or unbranched chains of Ubs. The combinations of substrates that can be ubiquitinated by the E3 ligating enzymes have only begun to be mapped (Suiter et al. 2025).
-
Countering ubiquitination are about 100 deubiquitinating enzymes with different specificities (Snyder and Silva 2021).
-
Ubiquitinated proteins are recognized by a plethora of Ub-binding effector proteins, triggering myriad physiological responses. Prominent among the effector proteins is the proteasome, which degrades the ubiquitinated proteins it recognizes (Ciechanover and Stanhill 2014; Grice and Nathan 2016); that indeed was the context in which Ub was first discovered. However, there are an unknown number of other effector proteins, mediating a largely unknown array of responses. Recognition of ubiquitinated proteins by effector proteins is primarily mediated by numerous small Ub-binding domains (UBDs), falling into 10 or 11 classes (Hurley et al. 2006). Effector proteins typically have multiple UBDs. Using Ub variants to probe specific effector functions is a major goal of this arena of investigation.
Affinity selection of Ub variants that bind specific elements of the Ub network is a promising new way to interrogate a great diversity of cellular functions. Binding of Ub-binding proteins to wild-type Ub is specific but weak; it seldom interferes with affinity selection of high-affinity ligands for specific Ub-binding proteins from libraries of Ub variants.
A project by Veggiani and his colleagues (Veggiani et al. 2022) exemplifies application of the principles outlined (see Overview: Generation and Characterization of Engineered Ubiquitin Variants to Modulate the Ubiquitin Signaling Cascade [Liang et al. 2024]). They prepared a panel of 60 different selectors, representing the Ub-interacting motif (UIM) class of UBDs (last bullet in the previous paragraph) from human proteins. Such UIMs bind weakly, but specifically, to wild-type Ub, with dissociation equilibrium constants in the range KD = 100–1000 μm. The selectors were used to affinity-select ligands from a library of Ub variants displayed on phages. The library was designed in light of the contact amino acids in the known structure of a Ub variant in complex with a UIM from yeast. Altogether 23 Ub amino acids were chosen for “soft” randomization, such that at each randomized position, ∼50% of the codons would encode the wild-type amino acids, while the other 50% would encode other amino acids. The resulting library contained 17 billion different Ub variants. A total of 138 distinct Ub variants, affinity-selected by 42 of the 60 UIM selectors, bound their selectors well above background levels. Further analysis of the most selective variant for each of the 42 selectors revealed high affinity and specificity for their cognate selectors:
-
Phage ELISA showed that 26% of the 42 variants recognized only their cognate selectors, and 52% recognized three or fewer selectors.
-
Competitive phage ELISA showed that 57% of the variants gave IC50 values <100 nm with their cognate selectors, while wild-type Ub gave IC50 values between 100 and 500 μm.
These results indicate that many UIMs, involved in a wide variety of physiological functions, can be specifically addressed with affinity-selected Ub variants. It seems likely that this approach will apply to UBDs other than UIMs as well. Design of a Ub variant library for each class of UBD can be guided by available structures of Ub in complex with members of that UBD class. A vast array of cellular effector functions would thus be opened up for detailed investigation.
The UIM project seems tailor-made for using HTS, rather than clone-by-clone analysis, as the first step in analyzing the output of affinity selections, as recommended above in “High-Throughput Sequencing (HTS) as Primary Readout in Affinity Selection Projects.” Thus, two rounds of affinity selection with all 60 UIM selectors would be carried out in parallel, stringency being greatly increased in the second round. HTS of the 120 first- and second-round outputs would allow clones of interest, with high specific affinity for their cognate UIM selectors, to be distinguished from the expected selector-unrelated phages (SUPs): phages that bind the carrier or other elements of the selection apparatus, phages that overreplicate during amplification, and phages that bind off-target UIMs. This line of experimentation would arguably be a streamlined way to choose a small number of clones for detailed individual investigation.
POSTSCRIPT: THE C.A.R.L.O.S. PROJECT
Carlos Barbas was one of the founders of the Phage Display Course, as explained above, and was a prime contributor to the phage antibody technology that is practiced worldwide in research laboratories and industry, and taught in the Phage Display Course to the present day.
In October 2013, Carlos sent an apology to the other instructors of the Phage Display Course. He would not be able to participate that year because he had just been diagnosed with metastatic medullary thyroid cancer (MTC). The terrible news galvanized Don Siegel at the University of Pennsylvania (Penn), codirector of the Phage Display Course from 1997 to 2019, to launch an emergency campaign to save Carlos's life. He recruited Christoph Rader, then still at Scripps, and Siegel's Penn colleagues Michael Milone and Vijay Bhoj to the cause. Here, I call them the MTC team and their campaign the MTC project.
Penn is a major center for development of a promising approach to cancers like Carlos's, called chimeric antigen receptors (CARs), in which some of a patient's own T cells are artificially hyperweaponized to eliminate the cancer. Milone, Carl June, and their coworkers had recently described a CAR (Milone et al. 2009) that would eventually be the first to be approved by the U.S. Food and Drug Administration (FDA) in 2017. It is now called tisagenlecleucel. Here, it is referred to as the anti-CD19 CAR because it targets a B-cell cell surface marker called CD19 that is also present on leukemia and lymphoma cells. The MTC plan was to develop a CAR that targets a cell surface protein marker (yet to be identified) on Carlos's cancer cells. They referred to the project informally as C.A.R.L.O.S.: Chimeric Antigen Receptor Lymphocytes with Oncolytic Specificity.
A CAR is an artificial T-cell receptor that mimics a native T-cell receptor (TCR); T cells expressing a CAR are called CAR T cells. Like a native TCR, a CAR is a membrane protein whose extracellular domain recognizes a specific antigen (protein marker) on the surface of target cells. A native TCR's extracellular domain recognizes specific peptide epitopes presented by MHC class I or II proteins on a target cell's surface. TCRs that recognize peptides from self-proteins are eliminated by immunological tolerance, severely limiting the repertoire of antigens that can be targeted. In contrast, the extracellular domain of a CAR is an scFv that binds directly to a specific surface antigen on the target cell. The repertoire of target antigens is not limited by tolerance. The scFv in the anti-CD19 CAR was constructed from a conventional monoclonal antibody. The antibody domain for the MTC project was to be a rabbit scFv obtained via phage antibody technology, once a target antigen had been identified.
In both TCRs and CARs, engagement of the extracellular domain with its specific peptide epitope or antigen on a target cell triggers a signal transduction cascade that activates the T cell, enabling several effector functions that result in death of the target cell. In the case of a native TCR, activation is triggered by accessory membrane proteins, including CD3 ζ, that are associated with the TCR noncovalently in a TCR complex. In the case of a CAR, the cascade is triggered by the intracellular domain of CD3 ζ, which is part of the CAR's intracellular segment. The intracellular segment also includes the intracellular domain of a costimulatory protein called 4-1BB, which triggers functions that enhance CAR T-cell persistence and performance (Philipson et al. 2020). A schematic diagram of the planned CAR's coding sequence is shown in Figure 9.
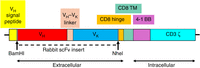
Schematic diagram of the planned chimeric antigen receptor (CAR)-coding sequence. This sequence encodes a rabbit scFv that specifically binds an antigen on the surface of Carlos's tumor cells. It is connected through a CD8 hinge and transmembrane (TM) to the intracellular domains of coreceptor protein CD3 ζ and costimulatory protein 4-1BB. The functions of the intracellular domains are explained in the text. CAR T cells (T cells expressing the CAR gene) deploy the CAR on the cell surface, with the single-chain variable fragment (scFv) exposed extracellularly, and the 4-1BB and CD3 ζ domains exposed intracellularly. Data are from Bhoj et al. (2021).
Producing the scFv component of a CAR for Carlos's MTC required identification of a suitable target surface antigen specific for MTC cells. Carlos had already arranged for a gene expression profile on a sample of his tumor. That profile was compared to a publicly available normal gene expression profile, pinpointing a few proteins that were overexpressed in the tumor. One of the overexpressed proteins was GFRα4, a receptor on the surface of MTC cells and their noncancerous precursors, but absent from almost all other cells in the body. The GFRα4 protein was used to affinity-select anti-GFRα4 Fabs from a 10-billion-clone naive rabbit Fab library supplied by Rader (Peng et al. 2017). The scFv form of one of those Fabs serves as the extracellular domain of the MTC project's CAR, which was constructed as described in Box 1.
Construction of a CAR and Preparation of Patient CAR T Cells
A chimeric antigen receptor (CAR) is constructed by inserting the coding sequence for the single-chain variable fragment (scFv) into the cloning sites (BamHI and NheI in the MTC project) of a lentiviral transfer plasmid, as diagrammed in Figure 9. The plasmid supplies the promoter and all elements of the CAR-coding sequence, except the scFv. Human producer cells in culture are transiently transfected with the transfer plasmid, along with a packaging plasmid encoding lentivirus proteins GagPol and Rev, and an envelope plasmid encoding the vesicular stomatitis virus (VSV) G surface glycoprotein. When the triply transfected producer cells are cultured, they release artificial enveloped virions with the structure of a lentivirus inside the envelope membrane, but with the VSV G protein deployed on the outside. The viral RNA molecules contain the CAR gene, but no viral genes. Lentiviral proteins GagPol (source of reverse transcriptase, integrase, and other proteins) and Rev are present as in a natural lentiviral virion, though without their genes. The artificial virions are harvested from the culture medium and frozen in aliquots for future treatment of patients.
To create patient CAR T cells, a preparation of the patient's T cells is infected with the artificial virions (the VSV G glycoprotein mediates infection of almost any cell type, including T cells). Inside an infected cell, the artificial CAR-bearing viral RNA is reverse-transcribed to DNA by the retroviral reverse transcriptase, with the help of the retroviral Rev protein. The resulting DNA is trafficked into the nucleus, and with the help of retroviral integrase is integrated into a random position in one of the T cell's chromosomes. The CAR gene in the integrated DNA is transcribed into CAR mRNAs, which in turn are translated to CAR proteins. The CAR proteins are trafficked to the cell membrane, with the scFv domain exposed extracellularly and the 4-1BB and CD3 ζ domains exposed intracellularly, as depicted in Figure 9. The virus cannot be propagated because no viral genes are present, and because the artificial CAR-bearing viral RNA is engineered to prevent the production of progeny artificial viral RNA by transcription of the integrated viral DNA.
The anti-GFRα4 CAR was tested in a mouse model of MTC (Bhoj et al. 2021). The “patients” were immunodeficient mice that would not reject engrafted human tumor cells. In the experiment described below, the mice were engrafted with cells of a human leukemia cell line that naturally expresses the CD19 surface protein, and that had been genetically engineered to express the GFRα4 surface protein and a luciferase enzyme in the cytoplasm. The purpose of the luciferase was to cause tumor cells to luminesce (emit bright light) when the luciferase substrate was administered to the mice, so the tumor could be imaged optically. The engrafted cells formed tumors within days. Five days after engraftment, the mice were infused with human CAR T cells expressing either the FDA-approved anti-CD19 CAR or the MTC project's anti-GFRα4 CAR, prepared as described in Box 1. As a negative control, mice were also infused with human T cells expressing no CAR at all. Two days before T-cell infusion (day 2) and on several days after T-cell infusion, groups of five mice were injected with the luciferase substrate, and 6 min later, the luminescence from their tumors was imaged. Figure 10 shows false-color images of tumor luminescent intensity, with blue for the least intense and red for the most intense.
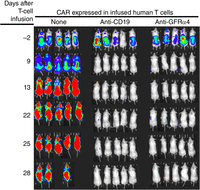
False-color images of luminescence from mice engrafted with a human tumor cell line expressing luciferase, and infused with human T cells expressing an anti-CD19 chimeric antigen receptor (CAR), an anti-GFRα4 CAR, or no CAR at all. False-color images of tumor luminescent intensity are shown, with blue for the least intense and red for the most intense. Adapted from Bhoj et al. (2021), under a Creative Commons License Attribution (CC BY-NC-ND); https://creativecommons.org/licenses/by-nc-nd/4.0/
The results in Figure 10 are striking: The tumors grew aggressively in the mice infused with the control T cells that did not express a CAR, but were rapidly eliminated in mice infused with human CAR T cells expressing either the FDA-approved anti-CD19 CAR or the anti-GFRα4 CAR developed in the MTC project. Based on these and other results reported in the article (Bhoj et al. 2021), the FDA has approved a Phase 1 trial of the anti-GFRα4 CAR in about 18 patients with metastatic or recurrent MTC (NCT no. 04877613).
Carlos will not participate in the trial. He died at age 49 on June 24, 2014, just as the mouse experiments were getting under way. It was only 8 months after his diagnosis. However, his legacy will live on—in the expanding realm of phage antibody technology to which he contributed, in the Phage Display Course he cofounded at the Cold Spring Harbor Laboratory, and perhaps in the anti-GFRα4 CAR if the FDA approves it for treatment of the cancer that cut short his life. Christoph Rader and two other colleagues of Carlos wrote a fitting appreciation of his many contributions to science (Rader et al. 2014).
COMPETING INTEREST STATEMENT
The author declares no competing interests.
ACKNOWLEDGMENTS
I am very grateful to Gregg Silverman, Don Siegel, Gianluca Veggiani, Brandon DeKosky, Mary Ann Pohl, and Juan Carlos Almagro for their generous help in writing this perspective.
Footnotes
-
From the Advances in Phage Display collection, edited by Gregg J. Silverman, Christoph Rader, and Sachdev S. Sidhu








