
Gateway-Compatible Yeast One-Hybrid and Two-Hybrid Assays
Abstract
In the first section of this introduction, we provide background information for yeast two-hybrid (Y2H) assays that provide a genetic method for the identification and analysis of binary protein–protein interactions and that are complementary to biochemical methods such as immunoprecipitation. In the second section, we discuss yeast one-hybrid (Y1H) assays that provide a “gene-centered” (DNA-to-protein) genetic method to identify and study protein–DNA interactions between cis-regulatory elements and transcription factors (TFs). This method is complementary to “TF-centered” (protein-to-DNA) biochemical methods such as chromatin immunoprecipitation.
PROTEIN–DNA AND PROTEIN–PROTEIN INTERACTIONS
Protein–DNA interactions (PDIs) and protein–protein interactions (PPIs) play pivotal roles throughout biology. PDIs are critical in gene transcription, DNA replication, recombination, and DNA repair. Gene transcription involves specific interactions between transcription factors (TFs) and cis-regulatory elements such as promoters and enhancers. Together, these PDIs form an integral part of the gene regulatory networks that instruct both organismal development and physiology (Walhout 2006). Approximately 5%–10% of all eukaryotic genes encode TFs (Reece-Hoyes et al. 2005; Kummerfeld and Teichmann 2006; Vaquerizas et al. 2009). To delineate gene regulatory networks, it is essential to identify the TFs that interact with all cis-regulatory sequences and to determine the DNA-binding specificities and affinities of individual TFs.
PPIs function in most if not all processes, from antibody–antigen interactions to signaling cascades that use multiple types of interactions for delivering information from the plasma membrane to the nucleus. Identifying proteins that interact with a DNA fragment or protein of interest can be highly informative about their function. For instance, proteins that physically interact with a TF may themselves be involved in gene expression; these proteins could be dimerization partners, cofactors, chromatin proteins, or proteins of the basal transcription machinery. Similarly, TFs that bind a DNA fragment that guides gene expression in a particular tissue may be involved in the development or physiology of that tissue. Several methods are available to identify PDIs and PPIs (for review of methods to identify PPIs, see Cusick et al. 2005).
Here we focus on genetic yeast “hybrid” assays that can be used in both small- and large-scale settings. Both yeast two-hybrid (Y2H) and yeast one-hybrid (Y1H) assays start with the generation of a “bait” that is used to “fish” for interacting “preys.” A cartoon for yeast hybrid assays is provided in Figure 1. The assays have greatly benefited from the development and application of the Gateway cloning system (Hartley et al. 2000; Walhout et al. 2000b) (see Introduction: Gateway Recombinational Cloning [Reece-Hoyes and Walhout 2018a]). Using the Gateway technique, many DNA fragments can be cloned in parallel, which greatly increases the throughput and lowers the cost of hybrid assays (Walhout et al. 2000a; Deplancke et al. 2004). After describing the concepts, methodologies, advantages, and disadvantages of Y2H and Y1H assays, we provide an associated set of protocols (see Protocol: Generating Yeast One-Hybrid DNA-Bait Strains [Reece-Hoyes and Walhout 2018b], Protocol: Generating Yeast Two-Hybrid Bait Strains [Reece-Hoyes and Walhout 2018c], and Protocol: Identifying Interactors from an Activation Domain Prey Library [Reece-Hoyes and Walhout 2018d]). Y1H and Y2H library screens are technically quite similar, except for bait generation and the different strains and markers used (see Fig. 2). Thus, we provide separate protocols for bait generation and a single protocol for library screens, with indications as to which media to use for which type of (Y2H or Y1H) screen. Finally, we discuss and provide protocols for Gateway cloning specific to these assays. The steps for Gateway cloning for Y1H and Y2H are shown in Figure 3.
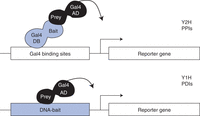
Principle of Y2H and Y1H assays. (Top) Yeast two-hybrid (Y2H) assays detect protein–protein interactions (PPIs). The “bait” protein is expressed as a fusion (or “hybrid”) with the DNA-binding domain (DB) of the yeast transcription factor Gal4, whereas the “prey” protein is expressed as a fusion with the Gal4 activation domain (AD). The DB-bait fusion protein can bind Gal4-binding sites artificially cloned upstream of reporter gene(s) that are integrated within the genome of the host yeast strain. Following an interaction with a prey protein, a functional Gal4 TF is created, and reporter gene expression is activated. (Bottom) Yeast one-hybrid (Y1H) assays detect protein–DNA interactions (PDIs). The “prey” is usually a transcription factor (TF) expressed as a fusion with the Gal4 AD, whereas the “bait” is a DNA fragment of interest. The DNA-bait is cloned upstream of reporters, and each DNA-bait::reporter cassette is integrated into the yeast genome. If the prey binds the bait, reporter gene expression is induced by the AD regardless of whether the prey TF is an activator or repressor in its native form.
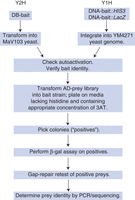
Pipeline for Y2H/Y1H library screening. This flowchart outlines the steps involved in setting up and performing Y1H/Y2H screens of an AD-prey library. Once the bait strains are generated, screening a library is technically very similar for both Y1H and Y2H.
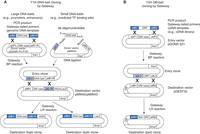
Overview of pipelines for Gateway bait/prey cloning for Y1H and Y2H. Gateway cloning uses recombination to transfer DNA fragments into plasmids. The direction of this transfer is dependent on the recombination enzyme and the recombination (or “att”) sites present in the plasmids. For instance, the BP enzymes recombine attB (black) and attP sites resulting in attL and attR sites, whereas the LR enzymes recombine attL (gray) and attR sites to generate attB and attP sites. Different variants of att sites are available that are only compatible with one another. For instance, attB1 only recombines with attP1 and not attP2. (A) Creating Y1H DNA-bait clones. DNA-baits can be amplified from genomic DNA using Gateway-attB-tailed primers (note that the att sites used for cloning DNA-baits are different from those used for ORF cloning in Y2H) and cloned into an Entry vector that contains attP sites using a Gateway BP reaction. This will generate an Entry clone in which the DNA-bait is flanked by an attL site and an attR site (and a by-product with the compatible attR and attL sites and the Gateway cassette; not shown). Small DNA-baits (up to ∼100 bp) can be cloned into the pMW#5 Entry vector by annealing oligonucleotides to generate a double-stranded DNA fragment containing the sequence of interest, with single-stranded overhangs that are compatible with restriction sites in pMW#5 (we routinely use HindIII and BamHI, but the pMW#5 multiple cloning region has sites for HindIII, SphI, SalI, BamHI, SmaI, and KpnI) (see Fig. 3 of Protocol: Generating Yeast One-Hybrid DNA-Bait Strains [Reece-Hoyes and Walhout 2018b]). The DNA-bait can then be transferred using a Gateway LR reaction from the Entry clone into two Destination vectors that contain attR and attL sites and either of the Y1H reporter genes (HIS3 or LacZ). (B) Creating Y2H DB-bait clones. ORFs can be amplified from a cDNA source using Gateway-attB-tailed primers and cloned into the pDONR 221 Donor vector that contains attP sites using a Gateway BP reaction. This will generate an Entry clone in which the ORF is flanked by attL sites (and a by-product with the attR sites and the Gateway cassette; not shown). The ORF can then be transferred using a Gateway LR reaction from the Entry clone into the pDEST32 Destination vector that contains attR sites. This Destination clone expresses the protein encoded by the ORF as a fusion with the DNA-binding domain (DB) of Gal4. These DB fusions are used as baits in Y2H assays. It should be noted that these ORF Entry clones can similarly be used to generate Destination clones in pDEST22 that generate a fusion with the activation domain (AD) of Gal4. Although the associated protocols specifically use libraries of AD-prey constructs cloned using conventional means, AD-prey clones generated using Gateway cloning can also be used as preys in both Y1H and Y2H assays. (GW cass) Gateway cassette (containing the toxic ccdB gene and a chloramphenicol-resistance gene), (ORF) open reading frame, (Kan) kanamycin, (Amp) ampicillin, (DB) Gal4 DNA-binding domain, (AD) Gal4 activation domain.
THE YEAST TWO-HYBRID (Y2H) SYSTEM: CONCEPT AND METHODOLOGY
The yeast two-hybrid (Y2H) system was developed by Stan Fields in 1989 (Fields and Song 1989). The conception of the assay was based on seminal observations in the transcription field, notably from work from Mark Ptashne and others who discovered that TFs can be composed of separable domains (Keegan et al. 1986). The yeast TF Gal4, which is used in Y2H and Y1H assays, is composed of a DNA-binding domain (DB) that specifically binds the Gal4-binding site with high affinity, and a transcription activation domain (AD) that is required for transcriptional activation of Gal4 target genes (Giniger et al. 1985). Both the DB and the AD can function independently of each other (Giniger and Ptashne 1987; Ma and Ptashne 1987). A fusion protein between the Gal4-DB and a heterologous AD (e.g., from the viral protein VP16) is capable of activating Gal4 target gene expression (Sadowski et al. 1988). Similarly, the Gal4-AD is fully functional when tethered to a promoter by protein–protein interactions (Ma and Ptashne 1988). Together, these observations paved the way for the development of yeast hybrid systems.
The Y2H system uses two hybrid proteins, the DB-bait and the AD-prey (Fig. 1). In the system for which we provide associated protocols, the DB and the AD are both from the yeast Gal4 protein. Alternative versions that, for instance, use the bacterial LexA DB and VP16 AD are also available (Golemis and Khazak 1997). The DB-bait is a hybrid protein composed of the Gal4 DB and the bait protein, for which one aims to identify or study interacting protein partners. The AD-prey is a hybrid protein composed of the Gal4-AD and the prey protein. Prey proteins can be obtained from (cDNA) library screens or by using individually cloned AD-prey clones. When the DB-bait and AD-prey are both expressed in the same yeast cell and if the bait and prey physically interact, a functional Gal4 protein is reconstituted and reporter gene expression is activated (Fig. 1). Integrated into the genome of Y2H-compatible yeast strains are cassettes with reporter genes downstream from Gal4-binding sites (Vidal et al. 1996b). DB-bait proteins can bind these Gal4 sites, and if an AD-prey fusion interacts with the bait, transcription of reporter genes is activated. Three reporter genes are commonly used in Y2H assays: the yeast genes HIS3 and URA3 and the bacterial LacZ gene (other systems are available that use other reporters). Expression of reporter genes is under the control of a basal promoter and a set of upstream Gal4-binding sites. The HIS3 reporter gene encodes an enzyme involved in the biosynthesis of histidine, and in its absence, yeast are dependent on the exogenous addition of histidine to the media. Similarly, yeast need a functional URA3 gene to grow in the absence of uracil. The strains used for Y2H assays carry deficient HIS3 and URA3 genes; alleles frequently used are his3Δ200 and ura3-52 (see Box 1 regarding selection of vector and yeast strain). Expression of functional HIS3 and URA3 genes can be selected using media lacking histidine and uracil, respectively. Additionally, levels of HIS3 induction are realized by adding the inhibitor 3-aminotriazole (3AT) to the media, such that growth in the presence of higher 3AT concentrations indicates higher HIS3 expression. The LacZ gene encodes β-galactosidase (β-Gal), and its induction can be measured by a colorimetric assay; in the absence of β-Gal, yeast are white, and in its presence, they turn blue. We have found that assessment of only HIS3 and LacZ activation in Y2H assays usually suffices. Having two reporters that both must be activated by the PPI increases the confidence in the relevance and/or robustness of such interactions (Walhout and Vidal 2001). Using the URA3 reporter can be helpful when interaction quality is difficult to judge or when the aim is to use reverse Y2H assays to identify interaction-defective alleles (Vidal et al. 1996a). In the latter, counterselection with 5-fluoorotic acid (5FOA) is used: An interaction confers an ability to grow on media lacking uracil but prevents growth on media containing 5FOA. If either the bait or prey protein carries a mutation that prevents interaction, colonies that grow on media containing 5FOA can be selected.
Choosing a vector and a yeast strain
When setting up a platform for yeast hybrid assays, the particular combination of vector and yeast strain can have a significant effect on the assay results. First, the yeast strain must be compatible with the assay. The strain cannot have functional GAL4 and GAL80 genes, which would interfere with the assay readout. The second consideration is the selectable markers used to determine that the yeast contain the vector(s) of interest. Almost universally, the selectable markers in yeast hybrid systems are auxotrophic growth genes that have been mutated in the original strain so that the yeast cannot grow in the absence of a particular molecule (e.g., an amino acid or nucleotide). This mutation is rescued when an ORF within the vector is expressed; for example, yeast mutated in TRP1 cannot grow in the absence of tryptophan unless transformed with the AD-ORF fusion vector that expresses wild-type Trp1p. In this way, the choices of vectors and of strain are interdependent because the strain must be mutated in the genes that are rescued by the desired vectors. Third, the user needs to consider the cloning and replication capability of the vectors. The majority of yeast assay vectors are available in both Gateway-compatible and traditional restriction-enzyme cloning versions. Although all vectors used in yeast contain sequences that enable their propagation in bacteria (e.g., ampicillin resistance and a high copy number bacterial origin of replication), their ability to replicate in yeast can differ. Integrative (YIp) vectors are not able to replicate in yeast and are preferred for plasmids that need to be integrated into the yeast genome by homologous recombination (such as the Y1H LacZ and HIS3 reporter constructs), because yeast transformed with these vectors, but not integrated, will not form colonies. YCp vectors replicate at low copy (one to three per cell) because of the presence of ARS (autonomously replicating sequence) and CEN (centromere) elements, whereas YEp vectors possess a segment of the yeast 2µ plasmid that serves as a high copy number origin of replication (10–40 per cell). Vectors that express the AD and DB fusion proteins are YCp or YEp vectors, with the YEp vectors making more of the fusion per cell simply because there are more copies. An alternative method for altering the amount of fusion protein is to use vectors with promoters of differing expression strength (e.g., a truncated ADH1 promoter drives less expression than the full-length promoter). Virtually, all AD and DB fusion vectors use the full-length ADH1 promoter. A final factor the user must consider is that different yeast strains simply behave differently in the assays. Many strains are available that are compatible with Y1H and/or Y2H, but it has been observed that some strains are more effective for identifying interactions than others. The best advice is to use proven strain–vector combinations or to test several combinations when instituting a new system.
In Y2H assays, the DB-bait first needs to be generated, and this requires a few decisions regarding experimental design (Table 1). First, one has to determine whether the full-length protein or only part of the protein (such as a conserved domain) will be used. This is important because not all interactions may be detectable with full-length proteins, and, conversely, interactions observed with protein fragments might not all occur in vivo. In addition, using protein fragments will require more screens per protein if a protein is to be fully assessed for interacting partners, but the benefit of this approach is that interaction domains are automatically uncovered (Boxem et al. 2008). Second, one has to decide on the vector to be used: one with an ARS/CEN or with a “2µ” origin of DNA replication. The former results in only a few copies per cell, whereas 2µ plasmids can result in many copies and can therefore lead to higher DB-bait protein expression. ARS/CEN plasmids may minimize false positives. However, the use of 2µ plasmids may lead to fewer false negatives (missed interactions). Third, one has to decide on the cloning method; one can use conventional restriction enzyme-based methods or, alternatively, one can use recombination-based methods such as Gateway cloning (see Fig. 3; Hartley et al. 2000; Walhout et al. 2000b). The latter is particularly desirable when multiple baits are to be cloned and screened. Other aspects of DB-bait generation can include the choice of promoter within the vector, which relates to the desired expression level of the DB-bait protein. Here, we describe DB-bait generation protocols by Gateway cloning, using ARS/CEN or 2 µ vectors. Our vectors use the ADH1 promoter to drive DB-bait expression.
Y2H DB-bait selection
After selection of the DB-bait, one has to decide how to identify interacting partners, that is, what source of AD-prey to use (Table 2). Again, there are several possibilities. The most widely used screens use high-complexity AD-cDNA libraries obtained either from a whole organism or from relevant tissues of interest (for this reason, the associated protocols only describe screening cDNA libraries). Most libraries derived from cDNA are not normalized, which means that a few clones are highly represented because they are expressed at high levels and in most or all cell types. As a result, many library clones need to be screened to identify less abundant AD-preys. To alleviate this issue, one can use ORFeome libraries that contain AD-ORF, rather than AD-cDNA clones. ORFeomes are defined as large collections of full-length open reading frames (ORFs), often cloned in a versatile (Gateway-compatible) vector (Reboul et al. 2001). Such resources are available—for example, for Schizosaccharomyces pombe (Matsuyama et al. 2006), Caenorhabditis elegans (Reboul et al. 2003), and human (Rual et al. 2004)—and these clones can be used to make either DB-bait (Fig. 3) or AD-prey constructs. In ORFeome-derived AD-prey libraries, all clones are represented approximately equally, so fewer colonies have to be screened compared with cDNA libraries. Additionally, these AD-preys can be screened in a directed manner, using either individual clones or pools of clones.
Y2H AD-prey selection
Using arrays of individual AD-prey clones ensures that all preys are examined for their capacity to bind the DB-bait independently (Uetz et al. 2000; Zhong et al. 2003; Grove et al. 2009). The AD-prey clones are placed at fixed coordinates in an array, introduced into the bait yeast strain in a parallel manner (by transformation or by mating), and interacting preys detected simply by phenotype comparison with negative control yeast containing AD-only plasmids. By this method, all the interactors present in the array are tested, and their identities are known based on their array position. However, although array-based screens using individual prey clones provide higher coverage than library-based screens, the applicability of this method to high-throughput experiments is limited by the availability of AD-prey clones and also by the size of the array. For instance, because it is technically challenging to screen an array that contains every isoform of every gene, only subsets of genes (e.g., TFs, kinases) are usually screened this way.
Pooling AD-prey clones is an effective way to interrogate many thousands of interactions. In this approach, the clone collection is subdivided into nonoverlapping pools of a limited number (tens or hundreds) and used for what are essentially small-scale library screens in which sequencing is used to identify interacting partners after the screen is completed. This method has been used to generate large “interactome” maps for C. elegans (Li et al. 2004) and human proteins (Rual et al. 2005). “Smart-pooling” places each AD-prey from the collection in multiple, strategically defined pools. All the smart pools are transformed into the DB-bait yeast in parallel, and computational deconvolution is then used to identify interacting partners from the positively scoring pools, thus reducing or even eliminating the need for sequencing (Vermeirssen et al. 2007b; Xin et al. 2009). With both types of pooling strategies, coverage is not as high as using arrays of individual clones, but it is better than library screens.
THE YEAST ONE-HYBRID (Y1H) SYSTEM: CONCEPT AND METHODOLOGY
The yeast one-hybrid (Y1H) system is very similar to the Y2H system, except that it uses a single hybrid protein, AD-prey, and a DNA fragment as bait (Li and Herskowitz 1993; Wang and Reed 1993; Deplancke et al. 2004). The DNA-bait is separately cloned upstream of two reporter genes, HIS3 and LacZ (see Fig. 3), and both constructs are integrated into the yeast genome (i.e., of the same yeast cell) (see Box 2). When the prey protein physically interacts with the DNA-bait, reporter gene expression is activated through the action of AD, and the readout is similar to Y2H assays. It is necessary to use prey fusions with AD because it enables the identification of both transcriptional activators and repressors, as well as proteins involved in other nuclear processes such as DNA replication (Li and Herskowitz 1993). Originally, the Y1H system used (multiple copies of) small sequences such as (predicted) cis-regulatory elements or TF binding sites as baits. However, single copies of larger and more complex DNA fragments such as promoters or enhancers can also be used (Dupuy et al. 2004; Deplancke et al. 2006; Vermeirssen et al. 2007a; Martinez et al. 2008a). In Y1H assays, two reporters are used: HIS3 and LacZ. The URA3 gene, which can be used as a reporter in Y2H assays, is used in Y1H assays as an integration marker for DNA-bait::LacZ reporter constructs. The DNA-bait::HIS3 reporter construct is selected on media lacking histidine. A minimal HIS3 promoter in the HIS3 reporter construct confers sufficient His3 expression to support growth. For the detection of PDIs, 3AT, which is a competitive inhibitor of the His3 enzyme, is included in the media. In the presence of 3AT, more HIS3 needs to be expressed to confer growth, and, hence, this compound can be effectively used in hybrid screens to identify PDIs.
Why integrate DNA-baits?
The primary advantage of integrating DNA-baits into the yeast genome is that it ensures a fixed copy number of the DNA-bait::reporters. This means that all yeast descended from a chosen integrant will possess the same number of reporter cassettes and will show a uniform level of background expression of the reporter. (Choosing the optimal integrant is discussed in Step 23 of Protocol: Generating Yeast One-Hybrid DNA-Bait Strains [Reece-Hoyes and Walhout 2018b].) In contrast, when replicative plasmids are used, the number of reporter plasmids present in each yeast cell can vary. Yeast cells with more plasmid copies may show higher background levels of reporter gene expression and can be mistaken for a positive in which a specific PDI occurs. A further advantage of integration is that the DNA-bait is chromatinized like the rest of the yeast genome. In this way, the DNA-bait is presented to the prey protein in a form that mimics in vivo conditions, rather than being presented as a “naked” plasmid in the cytoplasm.
Like Y2H assays, Y1H assays start with the choice and design of the DNA-bait (Table 3). There are two possibilities: You can use one or more copies of a small DNA sequence or a single copy of a larger, more complex DNA-bait. The former can be predicted cis-regulatory elements or TF binding sites, whereas the latter can be promoters, enhancers, or other complex genomic DNA fragments. We recommend a size limit of ∼2 kb for the latter, both to facilitate effective cloning and because longer-range PDIs may not be detectable in yeast.
Y1H DNA-bait selection
Broadly, the same issues apply for Y1H AD-prey selection as discussed above for Y2H assays. However, one important point is that Y1H assays are typically used to identify only TFs, and, therefore, small AD-TF minilibraries, smart pools, or clone arrays can be used and screens can be performed faster and more cheaply (Table 4; Vermeirssen et al. 2007b).
Y1H AD-prey selection
In sum, the choice of method for yeast hybrid screens depends on several factors: desired throughput, desired coverage, and perhaps most importantly, access to AD-cDNA or ORFeome resources.
Y2H AND Y1H ASSAYS: ADVANTAGES AND DISADVANTAGES
Y2H and Y1H assays have several advantages and disadvantages compared with other (biochemical) methods, specifically coimmunoprecipitation followed by mass spectrophotometry or chromatin immunoprecipitation followed by DNA sequence analysis (Table 5). The most important issue is that the assay is performed in yeast and not in the in vivo biological context (unless yeast proteins are used). This can be considered both an advantage and a disadvantage. The disadvantage is that follow-up analyses are required to determine the in vivo relevance/consequence of the interactions. The advantage is that the assays are largely condition-independent: As long as the interaction occurs in the yeast nucleus, these assays can detect proteins that would be difficult to retrieve by in vivo assays because they are expressed at low levels or in a tissue-restricted or temporally specific manner, for instance, during development or upon a physiological cue. However, these assays are not as effective with proteins that do not localize to the yeast nucleus (such as membrane proteins) or that require posttranslational modifications that do not occur in yeast. For such proteins, other hybrid assays may be more suitable, such as the split ubiquitin (Stagljar et al. 1998) or AVEXIS (Bushell et al. 2008) assays for membrane proteins, or MAPPIT (Eyckerman et al. 2001) or proximity ligation assays (Fredriksson et al. 2002) or LUMIER assays (Barrios-Rodiles et al. 2005) in mammalian tissue-culture cells for the detection of PPIs that depend on posttranslational modifications. Other advantages of hybrid assays include the detection of binary PPIs (Y2H), low-affinity or transient PPIs and PDIs (Y2H/Y1H), the use of Y2H to identify interaction domains (Boxem et al. 2008), and specific interaction-defective alleles (Walhout et al. 2000a); and the use of Y1H to identify TF binding sites (Reece-Hoyes et al. 2009) and novel TFs that do not have a recognizable DNA-binding domain (Deplancke et al. 2006; Vermeirssen et al. 2007a). Y1H assays are not yet adapted for the detection of heterodimers, a significant shortcoming because several TFs are known to bind DNA as obligatory dimers. However, homodimers are, in general, efficiently detected by Y1H.
Advantages and disadvantages of Y2H and Y1H assays
In sum, both Y1H and Y2H have advantages that make them powerful tools for finding and characterizing molecular interactions, but they also suffer from limitations that make some interactions undetectable (false negatives). False-positive interactions retrieved by these assays are discussed in more detail below.
FALSE POSITIVES
There are two types of false positives in hybrid assays: “technical false positives” that cannot be reproduced in the assay and “biological false positives” that can be robustly detected in yeast, but that do not occur in vivo. It is challenging to deem an interaction a clear biological false positive because assays that are used for in vivo validation can have their own limitations or false-negative rate. For instance, it may be difficult or impossible to validate interactions biochemically for low-abundant proteins or transient/low-affinity interactions. However, integration with other types of data can be very useful to add confidence to PPIs or PDIs. For instance, PPIs are more likely to be real when the protein partners are expressed in the same cell, in the same subcellular location, and at the same time.
When performed properly, the technical false-positive rate of hybrid assays is low (Venkatesan et al. 2009). There are several important issues to consider. First, the most important source of technical false positives is when the protein- or DNA-bait is highly autoactive, that is, it activates reporter gene expression in the absence of an AD-prey (see below). Some baits are “natural” autoactivators, that is, all the bait yeast show uniform high reporter activation (e.g., many TFs have their own AD and a few of these ADs function in yeast). However, some baits can turn into (i.e., de novo) autoactivators (Walhout and Vidal 1999), that is, a few individual yeast from a population with generally low autoactivity levels show high reporter activation due to mutations induced either by PCR (when initially cloning the bait) or during propagation in yeast. When spontaneous autoactivators occur in screens, they appear as positive colonies. However, when the AD-prey from that colony is retested with fresh bait-containing yeast cells (e.g., using gap-repair; for details, see Protocol: Identifying Interactors from an Activation Domain Prey Library [Reece-Hoyes and Walhout 2018d]), the interaction will not be reproduced. Thus, it is critical to retest PPIs and PDIs to avoid this type of technical false positives. In Y1H assays, it is critical to integrate the DNA-bait into the yeast genome to ensure fixed copy number and, thereby, fixed background reporter gene expression (Deplancke et al. 2004). In sum, technical false positives can occur but can usually be eliminated by repeated testing of interactions in fresh bait yeast cells.
PROTOCOLS FOR YEAST ONE-HYBRID AND TWO-HYBRID SYSTEMS
The associated protocols describe how to use Gateway recombination to generate bait constructs for both Y1H and Y2H assays, how to introduce these constructs into yeast to generate bait strains, and how to use these bait strains to screen for interactors from a library of plasmids that express AD-prey proteins. The processes for generating the constructs and strains are first described separately for Y1H (see Protocol: Generating Yeast One-Hybrid DNA-Bait Strains [Reece-Hoyes and Walhout 2018b]) and Y2H (see Protocol: Generating Yeast Two-Hybrid Bait Strains [Reece-Hoyes and Walhout 2018c]); another protocol describes how to screen a cDNA library (see Protocol: Identifying Interactors from an Activation Domain Prey Library [Reece-Hoyes and Walhout 2018d]). The latter is essentially the same for both Y1H and Y2H apart from using different selective media (indicated in Table 6). Constructs generated by traditional restriction cloning and ligation methods can also be used to create DNA or DB-bait and AD-prey constructs. Table 7 lists the vectors and primers used in the associated protocols.
Strains and markers used in Y1H and Y2H assays
List of vectors and related primers
Another three protocols provide supporting procedures that are used in each of the first three protocols (see Protocol: High-Efficiency Yeast Transformation [Reece-Hoyes and Walhout 2018e], Protocol: Colony Lift Colorimetric Assay for β-Galactosidase Activity [Fuxman Bass et al. 2016a], and Protocol: Zymolyase-Mediated PCR Amplification from Genomic and Plasmid Templates from Yeast [Fuxman Bass et al. 2016b]). These support protocols provide details on high-efficiency transformation of yeast, assaying β-galactosidase activity in yeast colonies, and yeast colony PCR.
ACKNOWLEDGMENTS
This work was funded by National Institutes of Heath grants DK068429 and GM082971 to A.J.M.W.
Footnotes
-
From the Molecular Cloning collection, edited by Michael R. Green and Joseph Sambrook.








