
Methods for Imaging Thick Specimens: Confocal Microscopy, Deconvolution, and Structured Illumination
Adapted from Live Cell Imaging, 2nd edition (ed. Goldman et al.). CSHL Press, Cold Spring Harbor, NY, USA, 2010.Abstract
When a thick specimen is viewed through a conventional microscope, one sees the sum of a sharp image of an in-focus region plus blurred images of all of the out-of-focus regions. High background, scattering, and aberrations are all problems when viewing thick specimens. Several methods are available to deal with these problems in living samples. These methods can be grouped into three classes: primarily optical (e.g., confocal microscopy, multiphoton microscopy), primarily computational (e.g., deconvolution techniques), and mixed (e.g., structured illumination) approaches. This article describes these techniques, which make it possible to see details within thick specimens (e.g., the interiors of cells within living tissue) by optical sectioning, without the artifacts associated with physically sectioning the specimen.
BACKGROUND
When a thick specimen is viewed through a conventional microscope, the depth of field (i.e., the distance between the top and the bottom of the in-focus region at a fixed setting of the focus knob) is <1 µm for the high-numerical-aperture (high-NA) objective lenses that are used for fluorescence microscopy. Thus, even when viewing a specimen as thin as 5 µm, 80% of the light may be coming from out-of-focus regions. The result will be a low-contrast image, composed of an intensely bright but very blurred background on which is superimposed the much dimmer in-focus information.
Thick and thin here refer to the thickness of the fluorescent material; overall specimen thickness per se does not increase the background. However, as the overall specimen thickness increases beyond 5–10 µm, other factors begin to degrade the image quality. When the illumination or imaging path intersects regions of widely different refractive indexes such as small granules or organelles, their curved surfaces act as microlenses to deflect the light in random directions. The consequence of multiple deflections may be to distort the light path enough to introduce aberrations into the image or even to scatter the light completely out of the field of view.
One way to eliminate the high background, scattering, and aberrations is to slice the thick specimen into many thin sections, which unfortunately requires fixation, dehydration, and embedding. That approach has limited application to the microscopy of living cells, but fortunately several other methods work well with living samples. These methods can be grouped into three classes: primarily optical (e.g., confocal microscopy, multiphoton microscopy), primarily computational (e.g., deconvolution techniques), and mixed (e.g., structured illumination) approaches.
Which Method to Use?
A systematic approach to choosing the best method is described at the end of the article, but a few introductory comments may be useful here. These methods are discussed here because they address problems encountered in imaging thick specimens such as living cells. For routine qualitative observation of relatively thin specimens (<3 µm), it will probably be much quicker and less frustrating to avoid them all and use conventional (wide-field) microscopy. However, there are a few situations in which the benefits of these more complex methods are important enough, even for a thin specimen, to warrant the extra cost, inconvenience, and time.
The most common application to thin specimens is when intrinsic contrast is very low, so that any loss of contrast, even the minimal decrease because of a small amount of out-of-focus light, complicates interpretation of the data. In this situation, all of these methods can usually improve contrast for any sample thicker than ∼2 µm. Another common application is to enable accurate measurement of the amount of a fluorescent component present in a cell, a task in which deconvolution methods excel. Finally, in the case in which a modest enhancement of resolution would change the interpretation of the data, then confocal, deconvolution, and some of the structured illumination methods are capable of delivering a small improvement over a conventional microscope. However, for most thin samples, the small improvement will not be worth the large extra effort. For very thin samples, <1 µm, quite extraordinary improvements in resolution are possible using several different modes of structured illumination (Heintzmann and Ficz 2007), but unfortunately these are not yet applicable to living cells.
For thicker objects that produce a moderate amount of out-of-focus light (typically 5–30 µm), any of the methods discussed here, and also multiphoton microscopy, should give a dramatically better result than a conventional microscope. When the sample is living (i.e., photobleaching and phototoxicity constrain exposure) and the signal is weak or the contrast is low, methods that must use photomultipliers for detection (e.g., point-scanning methods, confocal or multiphoton) have a severe handicap compared with methods that can use charge-coupled device (CCD) cameras (e.g., deconvolution, disk- or array-scanning confocal, structured illumination), because of the much higher quantum efficiency of CCDs. However, with very thick specimens that produce an overwhelming amount of out-of-focus light, only point-scanning (confocal or multiphoton) microscopy will give a satisfactory result.
How much is a moderate amount of out-of-focus light? Typically in such a specimen, the image seen through a conventional microscope will be too blurred to be useful, but one will be able to locate the region of interest and at least roughly set the focus level. Thus it is possible to locate the area to be imaged by visual observation, although the image will be too blurred to discern details. On the other hand, if the view through a conventional microscope is virtually featureless, giving no landmarks for choosing the appropriate area or for setting the focus, then currently the only choices are point-scanning confocal or multiphoton microscopy. These two methods can produce extremely useful images from outrageously bad specimens. However, from these very thick specimens, it is not realistic to expect a final image quality comparable to the best that a conventional microscope produces with a thin specimen, for reasons that we will consider in this article.
DECONVOLUTION METHODS
The goal of these techniques is to improve the images of thick objects by computationally removing the out-of-focus blur. The strategy is to calculate the structure of a hypothetical object that could have produced the observed partially focused image. The calculation is based on fundamental optical principles—in particular, a quantitative understanding of the effects of defocus—and may also take into account prior information or guesses about the specimen. The method commonly used is to refine iteratively an initial guess at the true object until the estimated image (i.e., the estimated object appropriately blurred by the effects of defocus) corresponds to the actual observed image.
Optical Principles
Successful application of these techniques requires an appreciation of how an image is formed by a microscope and what happens to an image when the lens is defocused. For this purpose, it is helpful to introduce the twin concepts of point-spread function (PSF) and contrast-transfer function (CTF). Both of these concepts describe the relationship between a real object and the image that is formed of it by an optical system. The PSF describes this relationship in terms of the image of a very small object, effectively a single point. Although the microscope can see objects as small as a single molecule, its limited resolution prevents the image from accurately portraying the size of very small objects, no matter what magnification is applied.
An illustration of this phenomenon is given in Figure 1. Notice that below a certain object size, images of every object appear the same. Increasing the magnification does not help; the image can be made larger, but not sharper. This limiting image is called an Airy disk, after the British astronomer G.B. Airy who first recognized its significance in 1834. Notice that, for this microscope, the Airy disk is not quite a perfect circle. The three smallest beads all appear to be slightly elliptical, with a weaker tail extending toward the upper right. Now, one might have been willing to concede that the manufacturing process somehow makes ellipsoids instead of spheres for all beads below a certain size, but it strains credulity to postulate that by coincidence, all of the beads that I photographed just happened to be oriented in exactly the same direction! In fact, electron microscopy shows that the beads are nearly perfect spheres. As Airy was the first to point out, the shape of this limiting image provides no information about the shape of the object—the Airy disk is an intrinsic property of the optical system itself. The Airy disk reveals that the image of a pointlike object is not a single point but is spread into a fuzzy disk. From this spreading is derived the more informative name for the Airy disk, which is the PSF. The PSF is often used as a means of quantitatively characterizing the performance of an imaging system.
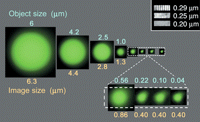
Epifluorescence microscope images of eight beads of known, decreasing size. The images of the fluorescent beads are shown in green, all adjusted to have the same maximum brightness. The actual brightness varied by ∼1000-fold. The true diameter of each bead is given above its image, and the diameter measured from the image is listed below it. The four smallest beads are shown at two different magnifications. The apparent diameters measured in the image are slightly larger than the true diameters, and the apparent diameters are the same for the three smallest beads, even though their true diameters differ by more than fivefold. These three images reveal the PSF of this microscope. The images in the upper right corner, obtained with the same microscope setup, show the appearance of three gratings with spacings of 0.29, 0.25, and 0.20 µm between the white bars. Notice that the contrast between the black and white bars decreases as the spacing gets smaller. In the actual object, the grating contrast is the same for all three scales. The bead images in the upper row are displayed at the same magnification as the gratings.
The grating images in Figure 1 show that the size of the PSF sets the resolving power of an optical system. An optical system forms an image by substituting its PSF for every point in the object and then sums all of these PSFs to make the image. The width of the PSF determines how far apart two points in the object must be to avoid being smeared together in the image. If the PSF of the optical system is broad, two points will have to be rather well separated to prevent the overlap of the two corresponding PSFs in the image. If their PSFs overlap extensively, then the two points will appear to be just a single point, smeared into a mushy average just like the lines in the image of the 0.2-µm grating. The smearing process is described mathematically as a convolution of the PSF with the object to produce the image. A good way to visualize this convolution is to imagine painting a picture of the object using a paintbrush with a tip the size of the PSF.
To take a concrete example, suppose that I had chosen not an individual 0.04-µm bead for the experiment in Figure 1 but instead inadvertently picked a pair of 0.04-µm beads separated by 0.08 µm, twice their own diameter. This pair would still be a smaller object than the single 0.22-µm bead, and their joint image would have the same shape as any of the three limiting images in Figure 1. In other words, 0.08 µm is well below the resolution of this microscope. It is important to realize however, that the Airy disk for the pair of beads would be twice as bright as the Airy disk from a single bead. In other words, the imaging process is a linear operation. The total intensity in an image of (A + B) is exactly the sum of the total intensity in an image of A plus the total intensity in an image of B.
A single number is often quoted for the resolving power of a microscope, such as the Rayleigh criterion, which is the radius of the Airy disk, given numerically by 0.6 λ/NA for incoherent imaging as in fluorescence microscopy, in which NA is the numerical aperture of the objective lens and λ is the wavelength of light that forms the image. However, using a single number for the resolving power is somewhat misleading because there is no sharp cutoff. As their size approaches the resolution of the imaging system, small details do not suddenly disappear. Instead, their contrast in the image becomes a smaller and smaller fraction of their true contrast in the object, until finally the image contrast approaches the size of the random fluctuations because of noise, and they then become invisible. The images of the gratings in Figure 1 show this progressive loss of contrast with decreasing size.
A complete description of the resolving power of an optical system thus requires information about the variation of contrast with size or, more precisely, the variation in the ratio of image contrast to object contrast as a function of size. The PSF actually contains this information, but it is revealed more clearly in its twin, the CTF. This function describes the extent to which contrast variations in the object are faithfully replicated in the image. Perfect contrast transfer means that image contrast equals object contrast. The CTF is usually expressed as a ratio, so that perfect contrast transfer means that the CTF has a value of 1.0. In the real world, things are less than perfect, and the CTF is always <1.0.
It is reasonable to expect that information about some features of the object might be transferred into the image more faithfully than others. For instance, the image may be a nearly perfect representation of the large-scale features of the object but contain much less information about the very smallest details. This will always be true for images obtained from an optical microscope because one cannot see clearly those details of the object that are small compared with the wavelength of light (i.e., details on the scale of the PSF). Thus the CTF is a function of the size of the feature being observed (Fig. 2, left). Normally the CTF is shown in a graphical form, plotting the ratio of image contrast to object contrast (vertical axis) against the reciprocal of size (i.e., spatial frequency). The CTF is simply a different representation of the same information given by the PSF of an optical system. Mathematically, the CTF and the PSF are related as Fourier-transform pairs.
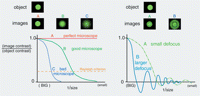
(Left) Schematic of some CTFs and the corresponding PSFs (object-image pairs for a very small object). (A) A perfect (impossible) microscope; (B) a typical good microscope; (C) a poor or improperly used microscope. The dashed line lies at a relative contrast of 25%, corresponding to the Rayleigh criterion for the resolving power of a microscope. (Right) Images of a tiny object from a microscope at two different values of defocus reveal the 3D nature of the PSF and CTF. Objects in some size ranges appear to change from black to white or vice versa as the focus level changes between the two indicated values. An example of this contrast reversal in a transmitted light image is shown in Figure 4.
Fundamentally then, when we speak of image resolution, we are, in fact, making a statement about image contrast at small spacings—alternatively, to be precise, about the ratio of image to object contrast at small spacings. Thus there is always an interaction between image contrast, image signal-to-noise (SNR) ratio, and image resolution, even though it is sometimes convenient to think of these as independent properties. What we normally refer to as visibility is determined by all three of those parameters and also by properties of the display system and the observer. Because of these subjective elements, visibility is not the last word—often one can extract much useful information concerning features that, by eye, are invisible.
The example of a typical good microscope CTF and PSF shown on the left-hand side of Figure 2 represents the case in which the specimen is thin and lies exactly in the focal plane of the objective lens. In fact, the CTF and PSF are three-dimensional (3D) functions. Their third dimension is revealed by comparing image to object when the object is displaced vertically from the focal plane of the lens. As the focus changes, a very surprising thing happens: Concentric rings appear in the PSF, and the CTF develops ripples, in some regions becoming negative. This means that for some features of the object, the image will have reversed contrast. For features in the size range corresponding to these negative oscillations of the CTF, dark parts of the object will appear bright in the image and vice versa (Fig. 2, right). As the degree of defocus increases, the CTF becomes increasingly oscillatory, with the contrast reversals affecting ever larger features in the image.
The image of a small fluorescent bead (i.e., the PSF) develops concentric rings as the lens moves away from focus (Fig. 2, top right). Changing the focus of the lens means that the 3D PSF is viewed at different levels along the optical axis. A vertical slice of the complete computed (Born and Wolf 1999, p. 489) 3D PSF, viewed from the side, is shown in Figure 3.

Vertical sections through the 3D PSFs calculated for lenses of two different NAs. The contrast has been greatly enhanced to show the weak side lobes (which are the rings of the Airy disk, viewed edge on). The small insets show the PSFs at their true contrast level. Note that vertical smearing (proportional to NA2) is much more strongly affected by the lens NA than is lateral smearing (proportional to NA).
It surprises most people that microscopes can produce such wildly incorrect images as depicted in Figures 2 (right) and 3. A striking example is shown in Figure 4, but the same effect can easily be observed on almost any specimen. In bright field, a small high-contrast feature such as a small dust particle or a scratch shows the contrast-reversal phenomenon clearly. Using a good dry or oil-immersion lens, carefully focus up and down by a small distance on either side of the correct focus, and you will see the particle oscillate from bright to dark and back again. If you can control the focus carefully enough, you may be able to find the position, halfway between a bright and a dark oscillation, where the particle becomes practically invisible (i.e., CTF ∼ 0 for details of this size). Evidently, some care is required in interpreting microscope images. How can you tell which is the correct appearance?

Contrast reversal because of defocus. (Left) A bright-field image of a diatom taken through a conventional microscope using a 60× (NA = 1.4) objective. (Right) An enlargement of a small portion of the image on the left, showing contrast reversals because of changing amounts of defocus. The diatom shell is curved, being thinner at the edges. As a result, the distance from the lens to the surface of the diatom varies; that is, the view includes a range of defocus values. Over this range, the CTF changes sign several times: From left to right, the holes change from black to white, to black again, and finally to white on the right-hand edge. The white rectangle indicates a narrow band midway between a black and a white hole region where the contrast of the holes is low; that is, the CTF is nearly 0 for structures of this size at this value of defocus.
To reiterate, a microscope substitutes its 3D PSF for each point in the 3D object and then sums all of those innumerable PSFs to give the final 3D image. The mathematical operation called convolution precisely describes this substitute and sum process. The distribution of intensities in the 3D image is the result of convolving the object-intensity distribution with a 3D PSF. The 3D PSF is an intrinsic property of the optical system and does not depend on the object.
Deconvolving Wide-Field Microscope Images
The 3D PSF (or equivalently, the 3D CTF) contains all the information one would need to predict the appearance of a known object when viewed through the corresponding optical system for any choice of focus. However, our problem is the converse of this. We know the appearance (i.e., the image), but we would like to know the real structure of the object. It is, in principle, possible to go backward in a one-step calculation from the observed appearance to the actual structure using the mathematical procedure of deconvolution. In practice, for realistic SNRs, a much better approach is to carry out this calculation in a multistep, iterative fashion. To see how the iterative deconvolution procedure works, imagine that the 3D object is made up of a stack of discrete two-dimensional (2D) planes. Normally the data will also be a stack of 2D-image planes, collected by changing the fine focus of the microscope by a small increment between successive images. To illustrate the iterative calculation, we will describe the steps for calculating one plane, say number 5, of an object that is nine planes thick (Fig. 5).

A simplified illustration of how one plane in the microscopic image of a thick object is formed. The process can be thought of as adding the in-focus image of one object plane to the images of neighboring object planes viewed at different amounts of defocus. For clarity, the process is illustrated for only two neighbors on each side of the in-focus plane. In reality, each image plane receives contributions from all object planes.
Consider plane number 5 of the observed stack of images (Fig. 5). When this image was recorded, what the detector saw was the sum of an in-focus view of object plane number 5, plus a view of object plane number 6 blurred by one increment of defocus, plus object plane number 4 blurred by one increment of defocus in the opposite direction, plus plane numbers 7 and 3 blurred by plus and minus two increments of defocus, respectively, and so forth. To simulate this process computationally, first make an initial guess at the real structure of the object on planes 1–4 and 6–9. Using the known CTF appropriate for each plane's defocus, blur these initial estimates, and add them together to estimate the contribution from out-of-focus blur to the observed plane number 5. Subtracting the sum of blurred object planes 1–4 and 6–9 from the image data for plane number 5 then gives us an estimate for the in-focus appearance of object plane number 5. Repeating these steps for all nine planes generates an improved estimate of the object. The entire sequence of operations on all planes is repeated in a loop until the object estimate no longer changes significantly (Fig. 6).
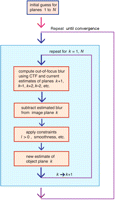
Flow chart for the constrained iterative deconvolution algorithm.
The description given above of the iterative deconvolution calculation is a simplified rendering of one of the early methods for deconvolving light microscope (LM) images (Castleman 1979; Agard 1984; Agard et al. 1989). Several other computational approaches have been reported (Erhardt et al. 1985; Fay et al. 1989; Holmes 1992; Carrington et al. 1995), and each of the commercially available packages incorporates its own additional proprietary features for improving the speed and accuracy of the convergence. These features include the application of various constraints at each cycle of the iteration. Smoothness constraints can be used, enforcing the physical impossibility of seeing intensity fluctuations on a scale much smaller than the known resolution of the microscope. Another possible constraint is the requirement that all values of intensity in the object must be >0. Some commercial packages used the so-called blind-deconvolution approach (Holmes 1992), in which the 3D PSF of the optical system is also estimated from the data to be deconvolved, instead of being experimentally measured from a separate 3D image of a small bead or computed from a theoretical model (Hopkins 1955; Stokseth 1969; Born and Wolf 1999).
Constrained iterative deconvolution is a computationally intensive task. It demands a significant computer power, and requires 1–2 min for a typical 3D dataset even on today's (early 2009) fast processors. Why would one choose this approach rather than the quicker and, at least on the surface, simpler approach of confocal microscopy? Not for better resolution: The resolution achieved by the two methods is comparable. Not for better background removal: With samples that are not outrageously thick, the two methods are approximately equal in their ability to remove the out-of-focus background light that degrades contrast. Where deconvolution plus wide-field microscopy is clearly superior to confocal microscopy is in the quality of the image data, measured as SNR (Murray et al. 2007).
There are several reasons why this is so, and these will be discussed later in the Interpreting the Results section of this article. The point to stress here is that with living samples, higher SNR becomes a top priority. If one is examining only fixed cells by, for example, labeling with antibodies, then quantitative measurements of the fluorescence intensity are not usually worthwhile because of the huge uncertainties inherent in immunocytochemical detection. However, it is now possible to image protein molecules in living cells. Suddenly, the types of questions one can ask have changed beyond recognition because the molecules can be directly counted (Femino et al. 1998). The yield of information from live cell experiments can be enormously increased if the SNR in the images is high enough to extract reliable quantitative estimates of fluorophore distribution (Swedlow et al. 2002).
Having said that, there are occasions, particularly if real-time evaluation is needed, when a quick-and-dirty contrast enhancement procedure is useful. For this purpose, there are several other less rigorous computational methods for enhancing the contrast in images that have been degraded by high background. It is very important to distinguish between true 3D deconvolution as described above, a mathematically linear operation, and the several varieties of fast, simple, deblurring algorithms (nearest neighbor, multineighbor, unsharp masking), which are fundamentally 2D operations, and mathematically nonlinear, although often cosmetically quite effective. Only the linear operation of 3D deconvolution restores image intensities so that they correspond quantitatively to the intensity distribution in the object. In doing so, this operation increases the SNR of the data (Fig. 7), and the output images are appropriate for all forms of quantitative measurement. The mathematically nonlinear deblurring algorithms can make the image look better, but the SNR is often degraded (Fig. 8). The output data from these nonlinear procedures may be acceptable for distance measurements on high-contrast objects, but are not usable for quantitative measurements of fluorophore distribution.

Deconvolution of wide-field microscope images of the parasite Toxoplasma gondii expressing YFP tubulin (Swedlow et al. 2002). (Left) One focal plane near the plasma membrane from a 3D stack of raw images of YFP fluorescence in several living parasites. (Middle) The same focal plane after processing of the 3D stack by constrained iterative deconvolution. Microtubules are clearly visible as bright striations. (Right) A plot of the change in intensity along the dashed lines in the raw data and in the data after deconvolution. Deconvolution greatly improved the SNR by restoring the out-of-focus light to its proper location. (Specimen kindly provided by Dr. Ke Hu, Indiana University, Bloomington. Images and deconvolution courtesy of Paul Goodwin, Applied Precision Inc.)
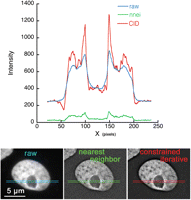
Quantitative comparison of 3D deconvolution with deblurring. A 3D stack of images of a DAPI-stained nucleus acquired on a wide-field microscope was processed by either full 3D constrained iterative deconvolution (CID) or deblurring using a nearest-neighbor algorithm (nnei). A single slice from the processed 3D stack, along with the corresponding unprocessed raw data, is shown. The graph shows the intensity profiles measured in a thin horizontal band across each image as indicated. Although both images have increased contrast relative to the raw data, note that the SNR is enhanced after 3D deconvolution but degraded with nearest-neighbor deblurring.
Practical Aspects and Tips for Generating Reliable Images
A superb guide to the practical aspects of deconvolving LM images has been published (Wallace et al. 2001). Anyone interested in using deconvolution methods will find that guide to be a gold mine of useful information. There is neither need nor space to repeat all of that information here, but a brief mention of some of the optical challenges in live cell imaging will be useful for the later discussion of confocal microscopy.
Modern objective lenses are nearly perfect when used under the conditions for which they were designed. Unfortunately, living cells, with all their structural variation and optical inhomogeneities (e.g., refractile blobs in phase contrast images), are far from the optical engineer's ideal object. Furthermore, when the goal is live cell imaging, a beautiful image of a dead cell loses to a mediocre image of a happy cell; when choices have to be made, optics is compromised to improve the cell's environment rather than the other way around. As a consequence, it is necessary to be able to recognize the more common forms of optical aberration (Cagnet 1962; Agard et al. 1989; McNally et al. 1994; Keller 1995) that afflict live cell imaging and to do what one can to compensate (Hell and Stelzer 1995). These aberrations are also discussed below, in the Confocal Microscopy and Interpreting the Results sections.
Spherical aberration is the manifestation of a difference in the focal position of paraxial rays compared with peripheral rays. It is usually induced by the presence of material with the wrong (i.e., not anticipated by the optical engineer) refractive index between the lens surface and the focal plane. For instance, using an oil-immersion lens to image a sample immersed in water will cause serious spherical aberration unless the sample is within a few micrometers of the coverslip. Use of a water-immersion lens avoids that particular problem (Fig. 9), but these lenses must be used carefully to avoid introducing other aberrations that interfere with deconvolution. Their performance is much more sensitive to the small tilt of the specimen, which induces a coma-like aberration (Arimoto and Murray 2004). When lenses designed for oil immersion are used with specimens mounted in water, the induced spherical aberration can be compensated by using an immersion oil with a refractive index higher than specified by the manufacturer (Hiraoka et al. 1990,) but only if the focal plane is <∼ 10 µm from the coverslip. However, the compensation is adequate only over a limited range of depths within the water and at the expense of introducing some chromatic aberration (Scalettar et al. 1996), because the dispersion of water is quite different from glass or immersion oil. Chromatic aberration is a manifestation of a difference in focal position and in magnification between light of different wavelengths. It causes a lateral and axial shift in the apparent position of objects of different colors, obviously a significant problem when one is trying to determine colocalization of different fluorophores. The apparent shift becomes worse with distance from the optic axis.
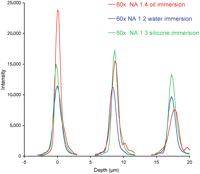
Effect of imaging depth on lens performance. Three-dimensional stacks of images of small fluorescent beads immersed in water were acquired, using an oil-immersion objective lens, a water-immersion objective, and an objective designed for use with a silicone oil as the immersion medium (refractive index ∼1.4). The graph shows the intensity of the bead image in each focal plane of the 3D stacks from three beads located at different distances from the coverslip surface (at depth = 0). For objects located immediately adjacent to the coverslip, the oil-immersion lens gives a brighter signal and sharper axial response than the other lenses. However, its performance is rapidly degraded by spherical aberration with increasing depth, whereas the other two objectives are less sensitive, so that at depths corresponding to typical eukaryotic cells the performance ranking is reversed.
The success of the 3D-deconvolution procedure depends critically on the accuracy of the image data and of the PSF. The inviolable rule of computation applies: Garbage in, garbage out. The most critical step of the deconvolution method is not the calculation but collecting the raw data. Computer software and hardware are the easy and cheap components of a quantitative 3D-imaging system. The expensive and difficult parts are the optical, mechanical, and electronic components that are required to collect precise, artifact-free, high SNR image data. Failing that, no amount of computation will help. The reliability of deconvolution depends absolutely on the reliability of the raw data used as input. To ensure this, the imaging conditions must be painstakingly optimized (Wallace et al. 2001), and there are several important preprocessing steps that must be performed to correct various artifacts typically present in 3D-epifluorescence image data (Hiraoka et al. 1990; Scalettar et al. 1996; McNally et al. 1999; Markham and Conchello 2001).
Limitations
As with any other technique, limitations to the use of deconvolution methods exist: Some specimens are unsuitable, and some suitable specimens challenge the currently available computational methods. One straightforward limitation is the need for a certain minimal SNR in the input data. All of the algorithms have the potential for amplifying noise. If the input signal is too noisy (i.e., the noise is large compared with the contrast between signal and background), then naturally the algorithms will fail, and the output will be meaningless. Effectively, this limits deconvolution methods to specimens in which the ratio of background fluorescence to in-focus signal is no greater than ∼20:1 (Murray et al. 2007).
Other limitations are not intrinsic to the method itself but are imposed by limited computational resources. For instance, the algorithms assume that the PSF of the optical system is the same for all points in the field of view (shift invariance) because the computations required to take account of a spatially variant PSF are not feasible for most applications (McNally et al. 1994). However, it is easy to show experimentally that this assumption is routinely violated in images of typical large (i.e., nonyeast) eukaryotic cells. Incorporating a measurement of local optical inhomogeneities based on differential interference contrast (DIC) imaging into an algorithm that allows for space-variant deconvolution is one promising approach to deal with this problem (Kam et al. 2001).
Another assumption that is incorporated into most algorithms (e.g., in smoothing filters that are used to constrain the intermediate calculations) is that the data are stationary in a statistical sense, which would require that the power spectrum be the same for every small region of the image. Obviously, this is far from true for any biological sample, particularly when the image records fluorophore distribution. Again, this assumption is not a necessary feature of the restoration algorithms (Castleman 1996, pp. 398–403) but a simplification to reduce the computational load. The practical consequence of violating this assumption is that small features can become unstable after a number of iterations, suddenly disappearing from the calculated result even though they may be present in the raw data.
A disadvantage of wide-field/3D deconvolution compared with, for instance, spinning-disk confocal imaging is the need to obtain a 3D stack of images even though the feature of interest may be confined to a single optical section. Phototoxicity caused by the extra exposure needed for the 3D stack may limit the overall length of time-lapse studies and may make the experiment impossible if fast processes are being studied. Even when the processes under study are relatively slow, such that an interval of many minutes between time points is adequate, cell movement on the timescale of seconds can nevertheless make it very difficult to acquire 3D data for deconvolution. An example is shown in Figure 10. Although no rapid processes were under study in that experiment, nevertheless the 2–3 sec required for acquisition of a 3D stack of images proved to be too long, because cell movements during the acquisition made reliable deconvolution impossible.
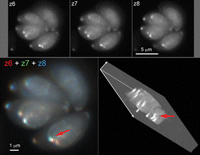
Movement artifacts in 3D image acquisition. The top row shows three adjacent planes from a stack of 17 focal planes of T. gondii expressing a GFP fusion protein that highlights the conoid and other structures. Small movements, difficult to appreciate in single images but obvious when the stack is replayed as a movie sequence, occurred during acquisition of planes 6, 7, and 8. Overlaying the three planes as red, green, and blue (RGB) channels of an RGB image reveals the movement of one conoid (red arrow). After deconvolution, viewing the 3D reconstruction at an oblique angle reveals the distortion (red arrow; jagged profile of the conoid, which is actually round).
Protection from being misled by these artifacts is of course purchased simply and in the same coin as for any other experimental undertaking: One must design and carry out sensible controls and repeat the experiment using independent methods and different conditions. Notwithstanding these minor difficulties, for suitable specimens, deconvolution of images from a wide-field microscope is a tremendously powerful technique that produces images of diffraction-limited resolution with less photobleaching and phototoxicity than any other method (Murray et al. 2007). It is an invaluable method that becomes increasingly important with the rapid progress in visualizing gene products in living cells.
CONFOCAL MICROSCOPY
The goal of this method is to improve imaging of thick objects by physically removing the out-of-focus light before the final image is formed (Minsky 1961; Petran et al. 1968; Brakenhoff et al. 1979; Carlsson et al. 1985; Amos et al. 1987). The method takes advantage of differences in the optical path followed by in-focus and out-of-focus light, selectively blocking the latter while passing the former on to the detector.
Optical Principles
Confocal microscopes differ from conventional (wide-field) microscopes because they do not “see” out-of-focus objects. In a confocal microscope, most of the out-of-focus light is excluded from the final image, greatly increasing the contrast and hence the visibility of fine details in the specimen. Figure 11 shows a comparison of images of the same thick specimen viewed by both wide-field and confocal microscopes. Figures 12 and 13 give schematics of the operating principle. On the left-hand side of Figure 12 is a wide-field microscope. A light source, in conjunction with a condenser, distributes light uniformly across the area of the specimen under observation. The diagram illustrates the paths followed by light arising from the specimen, passing up through the objective lens and eventually (ignoring some intermediate lenses that need not concern us here) reaching a detector of some sort: film, video camera, or retina. Three paths are shown, corresponding to light arising from three locations in the sample. The first location is in the center of the field of view and in the focal plane of the objective lens. The heavy dashed lines are the limits of the bundle of light rays that contribute to the image from this point. Similarly, the lighter dashed lines mark the rays from a second point in the same plane but displaced horizontally from the first point. Finally, light (the dotted lines), is coming from a third point located below the first point (i.e., from an out-of-focus plane). The light from this third point contributes to the blurred background, which we wish to eliminate from the image.

Images of a thick fluorescent specimen from a confocal and a conventional microscope. The sample is a chick embryo stained with propidium iodide and antibody against the carboxy-terminal glutamic-acid form of α-tubulin (fluorescein isothiocyanate [FITC] label). (Top left) Low-magnification, wide-field phase-contrast image of the entire embryo. The sample is ∼0.5-mm thick and contains a high density of refractile globules that scatter light efficiently. (Top right) Phase-contrast image at the same magnification as the fluorescence image. (Middle row) Conventional epifluorescence images showing (left) propidium iodide and (right) glu-tubulin distribution. The large amount of out-of-focus light severely reduces contrast. (Bottom left) Optical section obtained by confocal microscopy of exactly the same field and focal plane as the middle row. (Bottom right) Higher-magnification confocal view of a portion of the same field. Mitotic nuclei with condensed chromatin can be readily identified. (Dotted white ellipse) Bundles of tubulin are also seen. The mitotic spindle in these cells is formed predominantly with the tyrosinylated form of α-tubulin and hence is not seen. (Sample kindly provided by Dr. Camille DiLullo, Philadelphia College of Osteopathic Medicine.)
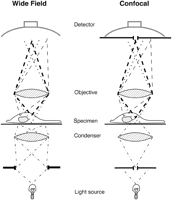
Schematic of the operating principle of the confocal microscope. (Left) A conventional, or wide-field, microscope. The specimen is illuminated over an extended region by a light source and condenser. Light rays arising from three points in the specimen are shown. The dashed lines emanate from two points in the focal plane, one centrally located (darker dashed lines), the other off axis (lighter dashed lines). The third point is on axis but located below the plane of focus (dotted lines); it gives a blurred image at the detector. The detector forms an image from the sum of all the simultaneously arriving light rays. (Right) A confocal microscope. Two pinhole apertures have been introduced. The upper aperture allows only the focused light rays from the on-axis, in-focus point of the specimen to pass to the detector. The lower aperture restricts the illumination so that it is focused on the point seen by the upper pinhole aperture.
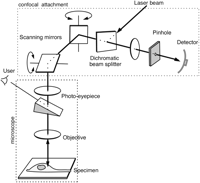
A typical laser-scanning confocal microscope. The instrument consists of a conventional fluorescence microscope (enclosed in the lower shaded rectangle) to which has been attached a confocal-scanning unit (upper shaded rectangle) comprising a pair of scanning mirrors, a laser, some wavelength-selective filters, a pinhole aperture, and a photomultiplier detector. The laser illumination is directed down the phototube of the microscope, having been deflected by the rapidly oscillating scanning mirrors so that it sweeps across the specimen in a raster pattern. Fluorescent light emitted by the sample passes back up through the phototube, is descanned by the scanning mirrors, and passes through the dichromatic beam splitter (which removes any reflected laser light) to the pinhole aperture. Light originating from the focal plane passes through the pinhole to the detector, but all other light is blocked. For reflectance imaging, the dichromatic beam splitter is replaced by a half-silvered mirror. A sliding prism allows visual (nonconfocal) observation through the usual binocular eyepieces, using the normal microscope lamps for illumination.
The right-hand side of Figure 12 shows how this is done, simply by adding a pinhole aperture to the wide-field microscope. Notice that behind the objective lens, all of the light rays are brought together at a crossover point, the location of the intermediate image plane of the microscope. Normally, the microscope oculars are focused on this plane to form the final fully magnified image. The location of this crossover plane along the vertical axis of the microscope is different for different light rays, depending on the distance of the corresponding point in the specimen from the front of the objective lens. The crossover point for light rays from the illustrated out-of-focus plane (dotted lines) is below that for rays from the in-focus plane (dashed lines). As illustrated, a pinhole aperture at the correct height will pass the converged rays from the in-focus point but block nearly all the dispersed rays from points higher or lower than the focal plane. (The geometry is slightly different in the so-called parallel beam confocal systems, but the principle is identical [Amos et al. 1987; Shao et al. 1991].) Out-of-focus points therefore contribute little to the final image; they are essentially invisible. An unfortunate side effect of the pinhole aperture is that most of the in-focus points also become invisible; only the rays from the central spot are passed by the aperture. We will see how to get around this problem shortly, but there is one more essential feature of the confocal microscope that we have to introduce first.
Because all of the specimen will be invisible except for the tiny spot imaged through the pinhole aperture, there is no need to illuminate an extended area. Illumination is needed over only a small area at any one time (i.e., the area that is visible to the detector looking through the pinhole aperture), and there are three good reasons for restricting the incoming light to this minimum necessary area. First, light going to other parts of the specimen will be scattered, and inevitably some of it will leak through the pinhole aperture, degrading the contrast in the image. Second, all of the illuminated area will be subject to photobleaching. Third, restricting the illumination to a single focused point gives a dramatic improvement in the discrimination against points above and below focus; in other words, it enhances the vertical resolution. The reason for this enhancement is as follows. If the incoming illumination is focused sharply to a point in the focal plane, then regions above or below this focal point will receive dispersed, much less intense, illumination. In fact, the intensity of illumination falls off as the square of the axial distance from the focal plane (i.e., intensity within the cone of illumination is inversely proportional to the cross-sectional area of the cone). Thus, when using this type of focused spot illumination in combination with the pinhole-blocked detector, not only will the pinhole aperture reject most of the light from out-of-focus planes, but also the light emitted from those planes will be less than it would have been with wide-field illumination. By exactly the same reasoning, the lateral resolution of the microscope will also be enhanced if a focused spot of illumination is used. These two modifications, limiting the area seen by the detector and limiting the area illuminated by the light source, are the key ingredients of a confocal microscope. A confocal microscope is simply an LM in which both the field of view of the objective lens and the region of illumination have been restricted to a single point in the same focal (confocal) plane (Wilson and Sheppard 1984).
To gain the optical sectioning capability of the confocal microscope, other aspects of the microscope's performance have been sacrificed. Field of view has been traded for increased axial resolution. The pinhole aperture effectively excludes light from out-of-focus planes, but it also restricts the field of view laterally to a spot the size of the demagnified pinhole. Thus, to gain the advantages conferred by the confocal pinhole, one must give up the convenience of acquiring an image from an extended area in parallel. The confocal image has to be built up sequentially by scanning one or more spots over the specimen until the region of interest has been covered (Fig. 14).
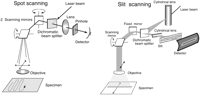
Comparison of spot-scanning and line-scanning modes of confocal microscopy.
Instruments
To make a useful image, obviously we need to see much more than one tiny spot of the sample. In principle, one could build up a complete image by scanning the specimen to and fro under a fixed spot of illumination or by scanning the objective lens, or the illumination, or the pinhole itself. In practice, because the scanning needs to be very fast to generate an image in an acceptable time, some types of scanning are much easier than others. There are four major types of confocal microscopes currently available, differing in the method they use to move the confocal spot relative to the specimen. In the simplest type, a single diffraction-limited spot is held stationary on the optical axis of the microscope while the specimen is moved (specimen scanning). The spot-scanning type uses a laser beam focused to a single diffraction-limited spot, which is deflected in a raster pattern over the specimen using oscillating mirrors or acousto-optical deflectors, and it uses a single fixed pinhole in front of the detector. In the disk-scanning type, a disk (Nipkow disk) containing an array of >104 tiny holes rotates in the illumination path, sweeping ∼1000 spots of illumination and conjugate detection pinholes over the specimen simultaneously. Finally, in the array-scanning (also known as swept-field) type, a stationary array of microlenses generates multiple beams that are swept across the field of view by a scanning mirror.
Specimen Scanning
There are important optical advantages associated with the stationary beam of the specimen-scanning type of instruments (Brakenhoff et al. 1979). All of the imaging takes place exactly on the optical axis, which minimizes many of the lens aberrations that plague the beam-scanning instruments. Alignment of the fixed optical path is also greatly simplified compared with systems with moving optical components. The primary disadvantage is that the specimen must be moved, along with the specimen holder, chambers, and (for living cells) liquid-culture medium. The total mass of the moving material is much larger than in a beam-scanning instrument, creating many opportunities for vibration and loss of positional accuracy. If the scan is to be completed within a reasonable time, the mechanical accelerations required are large, and only certain specimens are suitable. Even with the lightest specimens, the time resolution of specimen-scanning instruments is much worse than for other types of confocal microscopes and can be problematic for living samples. Sweeping a beam of light over a stationary specimen can be performed much more rapidly than moving a specimen under a stationary light beam. On the other hand, currently only a stationary-beam instrument can be truly confocal in transmitted light modes of imaging. The beam-scanning instruments are confocal only when the objective lens also serves as the condenser (e.g., epi-illumination fluorescence or reflection modes), for reasons that will be explained below. When the illumination and imaging light travel separate paths, as they do in all forms of transmitted light imaging (e.g., bright-field, phase-contrast, DIC), only the stationary-beam, specimen-scanning instruments are confocal.
Beam Scanning: Single-Spot Mode
In beam-scanning confocal microscopes (Fig. 13–15), the illumination is scanned while the specimen is held stationary (Carlsson et al. 1985). In the single-spot mode, a small (diffraction-limited) spot is swept over the specimen by means of a rapidly oscillating mirror interposed between the light source and the condenser lens (which is also the objective lens in epifluorescence mode). Because a useful image often consists of 105–106 pixels, the dwell time for each pixel needs to be kept very short to accumulate a useful image in a reasonable length of time. The need for fast scanning places stringent demands on the source of illumination because, of course, the number of photons collected per pixel also decreases as the dwell time is shortened. To collect a 512 × 512-pixel image in 1 sec, the scanning spot of light can dwell on each point for 4 µsec at most. In this time, one needs to collect as many photons as possible so that the statistical noise in the image is minimized. For this reason, a very intense source of light is needed, which in most instances means a small laser. A schematic of a typical laser spot-scanning instrument is shown in Figures 13 and 14. As illustrated, the laser is used as an epi-illuminator, as it is in most other types of confocal microscopes. The objective lens therefore also plays the role of condenser.
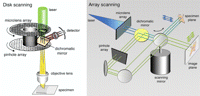
Disk-scanning and array-scanning modes of confocal microscopy.
Beam Scanning: Line Mode
A second form of beam-scanning microscope (Fig. 14) has been developed that is, strictly speaking, only partially confocal. These slit-scanning instruments replace the round pinhole aperture behind the objective lens with a long very narrow slit (Lichtman et al. 1989; Amos and White 1995). The illumination is shaped into a single narrow line, focused to the same line on the specimen that is seen by the slit aperture. Scanning is necessary in only one direction because the slit is long enough to admit light from points all the way across the field of view. The resolution and contrast in the image are no longer completely isotropic, but, in practice, the image quality is almost equal to that achieved with spot-scanning instruments for some types of sample, and the images are collected in a fraction of the time.
Tandem Scanning: Multiple Pinholes on a Nipkow Disk
In this type of confocal microscope, the spot of illumination and the detector pinhole move over the field of view in tandem. The same pinhole is used for forming the spot of illumination (on the input light path) and blocking out-of-focus light on the return path. Multiple spots are formed and imaged in parallel by means of an array of pinholes arranged in an Archimedean spiral on a rotating disk (Fig. 15, left).
The renaissance in optical microscopy that has been under way for the last two decades can, in some respects, be traced to the stir caused in the biological research community by the appearance of a remarkable new (an earlier invention had been reported but never developed [Minsky 1961, 1988]) type of microscope, a homemade tandem-scanning confocal microscope, and its equally remarkable Czech inventor, Mojmír Petráñ (Egger and Petran 1967; Petran et al. 1968; Egger et al. 1969). This small device (Dr. Petráñ carried it in his coat pocket) provided the first glimpse of the promise of confocal microscopy. It was a nightmare to align, but the images were astonishing and inspired the development of more user-friendly instruments (Boyde et al. 1983; Petran et al. 1986; Xiao and Kino 1987). Modern tandem-scanning microscopes are enormously improved from those developed in the early days and offer two important advantages compared to spot-scanning methods: First, the detector is a CCD (60%–70% quantum efficiency) instead of a photomultiplier (10%–20% quantum efficiency); and second, for some bright specimens, direct visual observation in real time is possible. At any one instant, only a few percent of the field of view is illuminated (1000 pinholes of the ∼25,000 on the disk), but the disk rotates fast enough that the moving spots fuse into a seemingly uniform image. Tandem-scanning instruments can, in principle, use broad-spectrum light sources such as high-pressure arc lamps for illumination. This would have the advantage of allowing a wider selection of fluorescent probes and would eliminate one source of noise that commonly contaminates images from laser-based instruments. However, in practice, conventional light sources are rarely used for fluorescence.
To avoid interference and maintain confocality, adjacent pinholes on the disk must be separated from each other by a distance that is large compared with their diameter. If they are positioned, say, 10 diameters apart, then only 1% of the incoming illumination will pass through the disk. To circumvent this inefficiency, one commercially available design uses a second disk, positioned above the pinhole disk, consisting of an array of tens of thousands of microlenses, each lens collecting light and focusing it on one pinhole (which are actually small transparent areas etched in the opaque coating of a clear plate), thus increasing the effective aperture of each pinhole (Fig. 15, left). With this design, ∼70% of the illumination light actually passes through the disk. Unfortunately, coherent laser illumination is nevertheless necessary. With conventional illumination, the focused spots from the lenslet array are much larger than the pinholes, again severely reducing the light throughput (Watson et al. 2002). A completely different design has been described that has efficiency comparable to the dual-disk lenslet array, in which a random mask of opaque and transparent patches, with 50% overall transmittance, replaces the array of pinholes (Juskaitis et al. 1996). Another approach that has been shown (Hanley et al. 1999; Heintzmann et al. 2001), and which one can hope will become developed to the point of commercial availability, is the replacement of the spinning disk with a stationary array of ∼106 individually addressable micromirrors on a chip (Texas Instruments digital micromirror device [DMD]), opening up a wealth of possible improvements over current confocal designs.
Tandem Scanning: Stationary Lenslet and Pinhole Arrays
Also now commercially available are two types of confocal microscopes in which multiple spots of illumination are generated by a one- or two-dimensional array of microlenses and swept over the field of view by a scanning mirror. The returning fluorescence is descanned using the same mirror to produce a stationary array of beams that is passed through a set of stationary pinholes. After the pinholes, the beams are rescanned so that they sweep over the surface of a CCD camera to form the complete field of view (Fig. 15, right).
Imaging Modes
Confocal microscopes can form an image using several different sources of optical information from the specimen. A reflectance image can be formed by using the light that is scattered (backscatter) from the specimen in the backward direction (i.e., back along the path of the incoming epi-illumination). This light will, of course, have the same wavelength as the original illumination. Colloidal gold labels are easily visualized in the reflectance mode. The insoluble precipitates formed by the Golgi stain procedure for neurons (Fig. 16) or horseradish peroxidase (HRP) oxidation of substrates such as diaminobenzidine also give bright backscatter images.
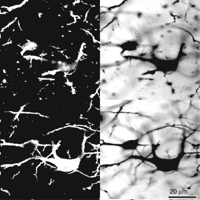
Confocal reflectance and nonconfocal transmitted light images of Golgi-stained neurons. (Left) Two optical sections made by using reflectance-mode imaging in a laser-scanning confocal microscope. The silver precipitate gives a very bright backscattered image. (Right) The corresponding nonconfocal transmitted light (bright-field) images.
In addition to the straightforward reflectance signal, the confocal microscope can also easily be set up to detect the interference pattern between light reflected from the cell membranes and that reflected from the underlying substrate (Sato et al. 1990), the technique known as interference reflection contrast microscopy (IRM) (Izzard and Lochner 1976; DePasquale and Izzard 1987, 1991). This method works well with intact living cells (Fig. 17). Fluorescence emitted by the specimen is the most common source of optical information used to generate confocal microscope images. In this case, the image-forming light has a different wavelength than the illumination, so the fluorescence and reflectance signals can be separated using dichromatic beam splitters as in conventional epifluorescence microscopes. A particularly valuable feature of the commercial instruments is the ability to acquire simultaneous, perfectly registered images from multiple different fluorescent labels with (in some instruments) independent control of the trade-off between sensitivity and resolution in each channel.

Confocal interference reflectance contrast imaging (IRM). Transmitted light DIC (left), single-wavelength (488-nm) IRM (middle), and dual-wavelength (488-nm, 633-nm) IRM (right) images of living fibroblasts from dissociated embryonic quail heart tissue growing on a glass substrate. In the IRM images, the contrast arises from interference between the laser light reflected from two surfaces (e.g., basal-cell membrane and glass substrate). The image was acquired at full aperture with a 63X (NA = 1.2) water-immersion objective, but the high degree of coherence of the illuminating laser light nevertheless causes the higher-order fringes (e.g., from interference between reflections off the apical and basal-cell membranes) to have much higher contrast than with conventional illumination (Izzard and Lochner 1976; Sato et al. 1990). The dual-wavelength IRM image is quite useful for discriminating between the zero-order (black) fringes, which occur at the same location for both wavelengths, and first- or higher-order (green, yellow, red) interference bands, which occur at different positions for the two laser lines. (Specimen kindly provided by Dr. Jean Sanger, Upstate Medical University, SUNY Syracuse.)
As well as forming an image from the emitted fluorescent light that is collected by the objective lens, most beam-scanning confocals have the ability to simultaneously collect illuminating light that passed through the specimen, acquiring a transmitted (e.g., bright-field, phase-contrast, or DIC) image in parallel with the backscatter or epifluorescence image. The quality of these scanned transmitted images is usually higher than that could be obtained using a conventional wide-field microscope, and they should be in perfect register with the simultaneously acquired fluorescence or reflectance confocal image. It is important to realize, however, that the transmitted light image is not confocal because there is no pinhole between the specimen and the transmitted light detector. Why not? The crucial difference between the transmitted and epifluorescent light is that only the latter encounters the scanning mirrors en route to the detector. Epifluorescent light from every point in the scanned field of view is reflected from the mirrors back along exactly the same path that the incoming laser illumination traversed: In other words, the epifluorescent light is descanned by the scanning mirrors and thus forms a stationary beam that can pass through a fixed pinhole. This is not so for the transmitted light. This light is not descanned and thus is not stationary anywhere along its path. In principle, one could introduce a second set of mirrors below the condenser, synchronized with the mirrors on the input side, to descan the exiting transmitted light. In practice, however, technical difficulties of this and other approaches (Goldstein et al. 1990; Art et al. 1991; Dixon et al. 1991; Dixon and Cogswell 1995) to descanning have so far blocked commercialization of a spot- or tandem-scanning instrument that is truly confocal in transmitted light.
Lasers and Fluorescent Labels
The total power required for imaging typical specimens is quite modest (∼0.1 mW) when compared with the power of commonly available lasers, but the intensity at the focal spot can be enormous (MW/cm2). It is important to use the minimum power necessary to acquire each image, which usually means reducing the beam intensity by 10- to 100-fold, using neutral density filters or an acousto-optic modulator in the illumination path. At very low laser power, the strength of the emitted fluorescence will increase directly in proportion to increases in the intensity of the illumination. However, as the illumination power is increased, the emitted light will no longer increase in proportion because the number of fluorophores already in the excited state becomes a significant fraction of the total fluorophores present. This phenomenon is referred to as ground-state depletion and should be avoided.
Increases of illumination power beyond the onset of ground-state depletion result in smaller and smaller increases in emission from the fluorescent molecules in the focal plane because they are mostly already in the excited state. Away from the plane of focus, where the illumination is less intense, increases in laser power will continue to excite more and more fluorescent molecules. This is an undesirable effect because the out-of-focus light does not contribute to the image and the excited molecules are subject to photobleaching. In this respect, multiphoton imaging has a possible advantage over confocal imaging because the light intensity is not high enough to generate multiphoton absorption events outside the focal spot. Unfortunately, many fluorophores photobleach faster with multiphoton than with single-photon excitation, so the advantage is sometimes not realized in practice (Patterson and Piston 2000).
Each type of laser emits light at a set of characteristic wavelengths, so the type of laser available determines the fluorophores that can be imaged. Table 1 shows the wavelengths available from some of the more common lasers and the major peaks in the spectrum from a mercury (Hg) arc lamp. It is important to remember that the peaks in the arc lamp spectrum are very much broader than the spectral lines from the lasers, and significant emission (5%–10% of peak intensity) occurs at all wavelengths between the peaks. Thus the range of fluorophores that can be excited by Hg arc illumination is very much broader than that for any single laser.
Visible laser and Hg arc emission wavelengths
Simultaneous Imaging of Multiple Labels
The distribution of wavelengths available from the light source becomes especially important when two or more fluorophores must be imaged in the same specimen. In general, one can expect problems with signal contamination between the two channels (bleed-through) when the emission ranges of two fluorophores overlap significantly and one of them is much more strongly excited than the other. For example, a rhodamine class plus a fluorescein class is one popular pair of fluorescent labels for double-labeling experiments in conventional epifluorescence microscopy, using Hg arc illumination at 495 and 546 nm. However, pairs of these fluorophores with spectra similar to rhodamine and fluorescein often give unsatisfactory results in confocal microscopes that use only an argon (Ar) or an argon–krypton (ArKr) laser because these do not emit appropriate wavelengths for efficient excitation of the rhodamine-class dye. Some instruments attempt to use the 488- and 514-nm lines of the Ar laser to excite the fluorescein class (typical peak excitation at 490 nm) and rhodamine class (excitation optimum ∼550 nm). Only the first fluorophore is efficiently excited at 488 nm, but it has an extended long wavelength tail of emission that completely overlaps the emission spectra of the second dye. At 514 nm, both dyes are excited equally (∼20% of maximum excitation). This combination of spectral properties and laser excitation wavelengths thus leads to severe problems with bleed-through. The problem is solved by using different fluorophores and/or different lasers. For example, the 488-nm Ar plus the 543-nm green helium–neon (HeNe) laser lines work well with these two dyes, or one can use dye number 1 plus a longer wavelength (Texas Red-class) dye using the Ar 488 nm and the 567 lines of the ArKr laser. For (much) more information, spectra, and an excellent discussion of applications of these and other fluorophores for live cell microscopy, the Molecular Probes catalog, or website (http://www.probes.com/), are invaluable.
As one increases the number of fluorophores used simultaneously, the probability of significant cross talk approaches 100%. There are several approaches to dealing with the problem, and new methods are introduced regularly. First, one can give up simultaneous data acquisition and instead use sequential scans with single laser lines. This works for combinations of fluorophores in which emission spectra overlap but that can be individually excited by different laser lines. It does not solve the problem when both emission and excitation spectra overlap extensively. An alternative is to greatly increase the spectral resolution of the detection apparatus (using a dispersive element of some kind, be it a prism, grating, or acousto-optic deflector), so that arbitrary wavelength bands of emission can be collected. Although it is sometimes possible to choose a narrow range of wavelengths that gives acceptable discrimination between fluorophores with partially overlapping excitation and/or emission spectra, the detected signal becomes weaker as the detection window is narrowed. One can also introduce additional criteria for gating the fluorescence output. For instance, fluorophores that have similar emission and excitation spectra may nevertheless have quite different fluorescent lifetimes. Very short-pulsed excitation and time-gated detection synchronized to the laser pulses then allow discrimination based on fluorescent lifetime (Gadella et al. 1993; Cole et al. 2001). Fluorescence polarization (Massoumian et al. 2003) (or hole-burning plus time-gated polarization-sensitive detection) offers additional possibilities for discrimination.
Inevitably, however, the number of signals that need to be separately detected will increase to exceed the capability of the detection system to discriminate, so ultimately one will have to deal with the problem of fluorophore cross talk by postacquisition processing. In this approach, multiple images contaminated with cross talk between different fluorophores are collected, each with a different combination of excitation and/or emission wavelengths. Reference images are also collected individually from pure samples of each fluorophore, using the same set of excitation and/or emission combinations. The contributions of each fluorophore are then sorted out by, in effect, setting up and solving the appropriate system of simultaneous linear equations for each pixel of the image. Applications of this computational method to karyotyping routinely discriminate between more than 20 different colors of fluorescent label (e.g., the spectral karyotype system from Applied Spectral Imaging) (Schrock et al. 1996; Ried et al. 1997).
Specimen Preparation
Confocal microscopy is compatible with any of the conventional specimen-preparation methods, including imaging of unprepared living tissue. Images at modest resolution of material down to a depth of ∼0.2 mm below the surface can be obtained from many tissues if the working distance of the objective is large enough. Thicker slices can often be examined completely if they are mounted between two thin coverslips and imaged from both sides. For the highest resolution work, spherical aberration introduced by the sample enforces a much lower limit on specimen thickness. Severe attenuation of both the incoming laser illumination and the exiting fluorescence emission, because of scattering by local inhomogeneities of the refractive index of the sample, often limits the quality of confocal images deeper than 0.05 mm. Attenuation is sometimes less for multiphoton microscopy, because of the longer wavelength used for illumination, and the fact that a detector pinhole is unnecessary, which gives multiphoton microscopy an advantage over confocal for deep imaging in some tissues. Care is necessary in mounting thick specimens to avoid compressing them while at the same time minimizing the distance between the coverslip and the specimen. It is also important to use small coverslips when possible. Large coverslips flex with each motion of the objective lens, causing fluid displacements and specimen motion.
Time-lapse imaging of living samples for extended periods at 37°C pose several problems. On–off cycling of the sample chamber heater results in vertical movements that shift the plane of focus. Because immersion lenses are usually required, the sample chamber will be strongly thermally coupled to the objective lens, requiring a separate objective-lens heater, whose cycling also changes the focal plane. In a typical microscope room, the on–off cycling of the laboratory air conditioner–heater system will cause additional focal shifts. However, with well-designed chambers and lens heaters and a modern laboratory building with good environmental stability, these thermally induced shifts are small enough to be barely noticeable under visual observation. Unfortunately, that is not good enough. Even under these ideal conditions, the shifts are still ∼10-fold larger than the vertical resolution of the confocal microscope (Fig. 18). To compensate for these shifts in unattended time-lapse imaging, the vertical extent of each 3D stack has to be increased both above and below the specimen by the size of the thermal shift. For instance, to be certain of completely capturing a 5-µm thick cell, images might have to be taken over a 15-µm span, tripling the total exposure and making it very difficult to do extended time-lapse imaging. A third problem arises when using water-immersion lenses. At 37°C, the water between the sample chamber and the lens quickly evaporates, and it is not possible to replace it without interrupting the time-lapse study and moving the specimen.

Homemade confocal microscope environmental chamber for live cell imaging. (Left) Front view of the temperature- and humidity-controlled box that encloses the entire inverted microscope except for Hg arc and tungsten lamps. Orange letters indicate the separate pieces of the box: (H) Ultrasonic humidifier (Vicks); (hc) relative-humidity controller (RHCN-3A, Omega Engineering); (T) heater–air circulator (Air-Therm, World Precision Instruments). (Middle) The disassembled pieces of the box, which are made of 6-mm acrylic sheet, that fit snugly together and are locked into place with clasps. The front piece (F) has a rectangular aperture through which the eyepieces protrude (orange rectangle). The front and right half of the top (RT) are clear acrylic. The left and right sides (LS, RS), left half of the top (LT), and back panel (B) are opaque, black acrylic covered with reflective thermal insulating bubble wrap. The back panel and floor plate remain permanently mounted on the microscope. The remainder of the box disassembles for use at room temperature. (Upper right) Top view from the right side, with the top of the box removed. The condenser/lamp-housing post of the microscope tilts backward for access to the stage, pushing a swiveling panel that is set into the back panel of the box. (Lower right) Graph showing focal-plane shifts with different sample chambers. Blue and green lines represent shifts with a commercial sample chamber and objective-lens heater in auto and confocal modes. The red line represents the shifts with the homemade box. After an initial equilibration period, the focal plane with the homemade box is stable to within ∼0.2 µm.
After struggling unsuccessfully with these problems, including trials with a wide range of commercially available sample chambers and temperature-control systems, we decided to take a different approach (Fig. 18). The specifications obtained from the microscope manufacturer revealed the fundamental problem. According to Zeiss, the overall thermal response for their Axiovert inverted microscope system, including lenses, stage, focus drive, etc., is 10 µm of focal-plane shift for 1°C temperature change. Therefore, to reduce the focal shifts to below the vertical resolution of the confocal microscope, it would be necessary to stabilize the temperature of the microscope to better than 0.04°C. That is not a realistic goal when the room temperature fluctuates by 1°C to 2°C, and the difference between the room temperature and the sample and/or lens temperature is 15°C–20°C. Moving the microscope to a warm room held at 37°C would be one solution, but the humidity needs to be very high to reduce the evaporation rate of the immersion water. A more user-friendly solution that has worked well for us is to enclose the entire microscope, except for the epi-illumination and transmitted light sources, in a (homemade) box held at 37°C and 70%–80% humidity (Fig. 18).
Photobleaching and Phototoxicity
In most cases, these two phenomena are actually the same process viewed from two different perspectives. When the emphasis is on accurate measurement of the 3D distribution of a fluorophore, then the primary concern is with photobleaching. When the emphasis is on observing the fluorophore distribution in a physiologically normal state, then phototoxicity will be the foremost concern. As a general rule, for quick observations of living cells at a single time point, photobleaching is the relevant phenomenon, and it is usually a surmountable problem. For repeated observations of the same cell, phototoxicity is the perspective that will be forced on us, because it is always a major problem and usually necessitates accepting compromises that limit image quality.
Modern confocal microscopes can acquire a high-quality digital image with much lower illumination than would be necessary for visual observation of the same sample. For example, Figure 19 shows the photobleaching of yellow fluorescent protein (YFP) in a living cell observed by confocal microscopy. In this specimen, as is often the case, the error because of photobleaching is small for a single image or even a moderately large 3D stack of images. Unfortunately, long before photobleaching makes the intensity measurement inaccurate, phototoxicity will have made the experiment irrelevant. To keep the damage to a minimum, careful attention must be paid to optimizing the microscopy. The goal is to extract the maximum information from the limited number of photons that the cell will tolerate before phototoxicity becomes unacceptable.
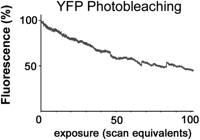
Loss of fluorescence of YFP because of photobleaching. Fluorescence from an YFP-fusion protein expressed in a living cell (a nondiffusible cytoskeletal component) was recorded using 514 nm excitation light in a laser-scanning confocal microscope. The illumination intensity, dwell time per pixel, and photomultiplier gain were adjusted to give an image of acceptable quality using a pinhole diameter equivalent to ∼1 Airy disk. The average fluorescence intensity in a small region was then recorded over the course of repeated scans. The fluorescence decreased by approximately one-half after 100 scans.
When the sample is not too thick, the much larger quantum efficiency of CCD cameras compared with photomultipliers gives disk-scanning confocals, wide-field, deconvolution, and structured illumination methods an important advantage over point-scanning confocal microscopes. For very thick living specimens where photobleaching is a serious problem, multiphoton illumination may provide some improvement, although this seems to be dependent on the sample (Patterson and Piston 2000).
Deconvolution of Confocal Images
Can deconvolution methods be applied to confocal images, and is anything gained by doing so? In principle, certainly. Although the confocal pinhole is said to remove out-of-focus light, this removal is never perfect, and unquestionably confocal images always show residual effects of the ripples in the 3D PSF caused by defocus. An example is shown in Figure 20, a confocal reflectance image of the same diatom imaged by wide-field microscopy in Figure 4. Notice that the phase reversals are seen in the confocal image; the holes change from black to white across the curved diatom surface. This suggests that confocal images might benefit from deconvolving the 3D PSF, and indeed this has been reported (Shaw and Rawlins 1991; Cox and Sheppard 1995; van der Voort and Strasters 1995; Verveer et al. 1999; Boutet de Monvel et al. 2001). However, there are several considerations that might make investigators wary of the results, and several more reasons why they might conclude it is not worth the effort.
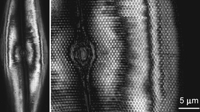
Contrast reversal because of defocus in a confocal image. (Left) A reflectance image of a diatom taken with a confocal microscope (Olympus FluoView 300) using a 60× (NA = 1.4) objective. (Right) An enlargement of a small portion of the image on the left showing contrast reversals because of changing amounts of defocus. Compare with Figure 4.
The first challenge is to determine the correct 3D PSF for a confocal microscope. In theory, this can be computed (Wilson and Sheppard 1984), but in practice, the computation is inaccurate, as it is for conventional wide-field microscopy with high-NA objectives. Can a 3D PSF be experimentally determined, as is routinely performed for wide-field microscopes? Unfortunately, the images from confocal microscopes often suffer from patterned noise artifacts (Fig. 21). Some of the patterns repeat with a periodicity that is a significant fraction of the entire field of view, which means that they are not represented at their true contrast in the images of tiny beads that are used for PSF measurements. Effectively, the PSF varies across the field of view, contrary to the assumptions of typical deconvolution algorithms. A second concern is the poor SNR of confocal images (Figs. 21–23). This makes it quite difficult to measure the (local) 3D PSF with the accuracy required for reliable deconvolution and greatly exacerbates the tendency of deconvolution to amplify noise in the raw data.
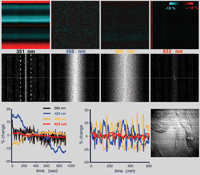
Fluctuations in illumination intensity and polarization in a confocal microscope (Zeiss LSM 510). Mean-corrected images of a uniform specimen (top row) and their power spectra (middle row) acquired with four different laser lines. The experiment was performed under conditions that gave >1000 photons per pixel, and thus the expected single-pixel noise in the images is ∼3%. The images would ideally be of uniform intensity and thus be entirely black after subtraction of the mean value but instead show artifactual intensity fluctuations. Deviations from the mean value of up to ±10% are color coded according to the scale in the top right corner. The power spectra show that each laser contributes patterned noise with a complex mixture of periodicities. Contrast of the power spectra has been enhanced to make the weaker features visible in the print. Some of the patterns have repeat lengths that are much longer than a single scan line. (Bottom row) The average fluctuation in intensity in a 100 × 100-pixel image of a uniform, stable fluorescent sample is plotted for a series of 1000 images acquired at 1-sec intervals (left), or 600 images at 1-min intervals (middle). The expected noise in the average value of 104 pixels is ∼0.03% (i.e., 0 with 8-bit intensity digitization). The effect of these artifacts is to severely degrade the image SNR (see Figs. 22 and 23). The horizontal stripes in the DIC image (bottom row, right) are caused by random changes in plane of polarization that accompany the fluctuations in intensity of the illumination.
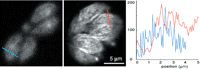
Spot-scanning confocal images of transgenic Toxoplasma gondii expressing an YFP–α-tubulin fusion protein. Microtubules near the cell surface are included in these single optical sections. (Left) A typical spot-scanning confocal image. (Middle) A superior image (Olympus confocal, much less intensity fluctuation); the best ever recorded from this specimen. Compare the SNR in this image with the wide-field image in Figure 7. (Right) A graph showing the intensity profile along the red and blue lines in the images.
The number of photons needed to give a maximum signal (gray level = 255) on a typical spot-scanning confocal microscope as a function of the gain setting (PMT voltage). On this microscope, typical specimens require a gain setting of 800–900 V, corresponding to ∼180–80 photons/pixel at maximum brightness.
These concerns apply even to images of thin specimens. If a specimen is thick enough that one really needs to use a confocal microscope (instead of using wide-field plus deconvolution microscopes), then the 3D PSF is certain to be seriously degraded by spherical and chromatic aberration (Figs. 24 and 25), and this distortion will change dramatically between the top and bottom of the 3D data stack. In this situation, deconvolution is unlikely to give a correct result.
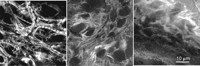
Confocal optical sections of thick tissue. The developing heart in a chick embryo, labeled with fluorescent antibody against cardiac myosin, was imaged with a 60× (NA = 1.4) oil-immersion objective. (Left) The first optical section of a 3D stack of 160 sections spaced at 0.5-µm increments. The first section was ∼20 µm below the coverslip. (Middle) Section number 30; (right) section number 130. Spherical aberration and scatter increasingly degrade the resolution, so that the 2.2-µm myofibrillar striations, clearly visible in section number 1, are barely resolved in section number 130. (Sample kindly provided by Howard Holtzer, University of Pennsylvania.)
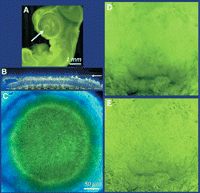
Comparison of laser-scanning confocal microscope and a wide-field microscope performance in imaging a thick tissue (Swedlow et al. 2002). A 5-d-old quail embryo stained with Alexa 488 phalloidin (green) and DAPI (blue) was imaged by laser-scanning confocal (A–C) and wide-field microscopy (D). (A) A low-magnification survey image of the entire embryo. The head is at the top of the figure. The arrow points to the developing eye, the region shown at higher magnification in B–E. (B) x–z section (parallel to optical axis) showing position of the coverslip (arrow) and the location of the focal plane shown in C (dashed line). (C) A single optical section ∼50 µm below the exterior surface of the embryonic eye. Concentrations of actin at cell cortices are visible. (D) The same embryo imaged by wide-field microscopy and recorded with a CCD camera (Alexa 488 only). The image shown is one of the 60 recorded from a series of optical sections. (E) The same optical section as in D after restoration by deconvolution. No cellular details are visible in wide-field images of this thick tissue. Scale bars: (A) 1 mm; (C, also for B,D,E) 50 µm. (Images D and E courtesy of Jason Swedlow, University of Dundee.)
Finally, it could be reasonably argued that deconvolution comes too late to correct the most important defects of confocal images. The great benefit of deconvolving wide-field images is that the SNR is enhanced because at least some of the out-of-focus light can be restored to the in-focus plane, thus increasing its total information content. In a confocal image, however, virtually all of the out-of-focus light was thrown away (blocked by the pinhole) before reaching the detector and thus cannot be retrieved by deconvolution. This removes much of the motivation for applying deconvolution methods to confocal images.
Practical Aspects and Tips for Generating Reliable Images
The currently available confocal microscopes are rather delicate, unstable instruments. They are typically controlled by complex computer programs and are, therefore, prone to unexplained crashes and failed operations or missing functions. For these reasons and more, acquiring high-quality confocal images that faithfully represent the sample is a slow, often frustrating process, and the path to success is sprinkled with traps for the unwary. Unfortunately, even with a complete novice at the controls, an image of some kind will always appear on the screen. However, distinguishing image from garbage takes time and considerable care regardless of how much experience the operator has.
Below are some guidelines that may help in adjusting the microscope parameters to obtain interpretable confocal images. (Additional help can be found in the Troubleshooting Guide.) For the preliminary adjustment of imaging parameters, choose an area of the specimen that is roughly equivalent to the area that will be recorded but is not the best area. The chosen area will be destroyed during the setup phase.
-
1. Choose the appropriate combination of laser, dichromatic mirror (beam splitter), and emission filter (see comments above relevant to imaging multiple fluorophores).
-
2. Decide what pixel spacing is appropriate for collecting the information you need from this particular sample, and set the magnification or electronic-zoom factor accordingly.
-
Do not oversample (the pixel spacing should be only slightly smaller than the Nyquist criterion, i.e., slightly less than one-half of the spatial resolution required for the experiment). Use the highest-NA objective available. For thick samples mounted in aqueous media, a water-immersion objective with a correction collar set to minimize spherical aberration is the best choice (see Interpreting the Results, below). For thinner samples, an oil-immersion lens may work, but the refractive index of the immersion oil must be carefully selected to minimize the spherical aberration for each specimen.
-
-
3. Get a ballpark estimate for the imaging parameters.
-
i. Set the pinhole initially to ∼1 Airy disk unit.
-
ii. Set the laser to the minimum power that gives a decent signal at maximum gain on the detector.
-
-
4. Find the linear range of the detector system: Use a pseudocolor lookup table (LUT) that highlights underflow (intensity = 0) and overflow (intensity = 255 for 8-bit or 4095 for 12-bit detections) in color, but is grayscale in between.
-
i. Scan with the laser off or set to zero power at the scan speed to be used for the specimen, and adjust the offset (dark-current compensator) so that the recorded image intensity is minimized but there are no pixels at zero intensity.
-
ii. Find a region of the specimen that is likely to be the brightest and, with the laser on, decrease the detector gain until the recorded intensity in the brightest region of the image is safely below the saturation value (e.g., 200 out of the maximum of 255 for an 8-bit system).
-
-
5. Find the linear range for the specimen: Check that the recorded fluorescence emission increases linearly with an increase in laser power up to at least twice the laser power you will use for imaging.
-
If the emission does not increase in proportion to the laser power (i.e., ground-state depletion is occurring), you will have to sacrifice time resolution (work with lower laser power and longer scan times) or spatial resolution (work with lower laser power and increased pinhole diameter or increased pixel size) or both.
-
-
6. Check that you are below the instantaneous damage threshold. With the scan speed and laser power set at these preliminary values, monitor the image intensity in a selected small area of the specimen over the course of numerous repeated scans.
One would like to be able to scan dozens of times before the cumulative photobleaching reaches 50%. If the fluorescence is bleaching too much, you will have to sacrifice either temporal resolution (longer intervals between scans in a time-lapse series) or spatial resolution (increased pinhole diameter and spacing between optical sections; increased xy pixel size) or both. If the fluorescence is not bleaching measurably and it is to be a quick experiment or you are otherwise certain that photodamage is not a concern (lucky you), then you may decrease the photomultiplier gain (which will decrease the noise) or decrease the pinhole size (which may improve contrast and resolution) while increasing the laser power to maintain maximum intensities just below saturation.
-
7. Iteratively readjust the parameters according to Steps 4–6 until the image SNR is optimized.
Limitations
The most important limitation of confocal microscopes compared with wide-field/3D deconvolution is the lower SNR of confocal images, a consequence of the much lower photon efficiency of confocals. In a side-by-side comparison, using a very thin specimen with no fluorophores in out-of-focus planes, using the same lenses, same filters, same wavelength, and equal illumination dose, a typical wide-field microscope will collect two to four times more photons in the image than a spinning-disk confocal microscope. The efficiency of spot-scanning confocals is lower still; ∼50-fold fewer photons will be collected in the image than with a spinning-disk confocal microscope, and several-hundred-fold fewer than a wide-field microscope at an equal illumination dose. The lower quantum efficiency of the photomultiplier tube (PMT) detectors on spot-scanning confocals compared with the CCD detectors on a spinning-disk or wide-field microscope (∼15% vs. ∼70%) accounts for some of this lower efficiency but clearly not all. The other reasons for these dramatic differences in efficiency are unclear at present, but they are observed in all machines tested and thus seem to be intrinsic properties of the different optical designs rather than examples of poor engineering in any one brand (Murray et al. 2007).
As a result of this low efficiency, the number of photons per pixel in a typical image from a spot-scanning confocal is astonishingly small (Fig. 23), rarely >100 and often <20. Obviously, with 20 photons/pixel and consequently ∼20% intensity fluctuation because of Poisson statistics alone, spot-scanning confocal images will have limited usefulness for quantitative measures of fluorescence. Spinning-disk confocal images are somewhat better (Fig. 26) but still not of the same quality as deconvolved wide-field images.
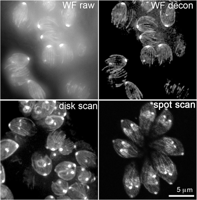
Images of Toxoplasma gondii expressing YFP-tubulin. (Top left) The raw wide-field (WF) image, (top right) the result obtained after deconvolution, (bottom left, bottom right) images obtained from different regions of the same sample using spinning-disk and spot-scanning confocal microscopes, respectively. The same specimen was imaged on all three microscopes using equivalent objective lenses and fluorescent filters. In all cases, the images shown are the sum of three adjacent focal planes spaced 0.3 µm apart. The cortical microtubules are visible as lines of intensity and the conoid as a bright dot at the apical end of each cell.
In comparison to spot-scanning confocals, disk-scanning and array-scanning confocals have more limited ability to remove out-of-focus light because of cross talk between the multiple pinholes. The situation is shown schematically in Figure 27, a diagram of the light distribution in a multiple-pinhole confocal. Overlap of the region of acceptance from adjacent pinholes restricts the specimen thickness from which out-of-focus light is excluded. An experimental demonstration of this effect is given in Figure 28, showing the integrated intensity recorded from a single point as a function of defocus, for wide-field, spinning-disk, and spot-scanning confocal microscopes. Note that the spinning-disk microscope performs identically to the spot-scanning confocal microscope within ∼1 µm of the focal plane, but beyond that the light from the out-of-focus bead is no longer excluded.

Schematic of the light distribution in a multiple pinhole confocal microscope. (Top left) A vertical slice (i.e., x–z plane) of the distribution of illumination near the focal plane for a single spot of illumination with an objective lens of NA = 1.2. (Bottom left) The corresponding fluorescence emission with the acceptance angle of the objective superimposed. (Right) The overlap of neighboring illumination cones and acceptance regions, with a spacing between adjacent spots/pinholes of d. This limits the region of confocal behavior—that is, the ability to exclude light from out-of-focus planes—to a narrow band on either side of the focal plane. Out-of-focus light that originates from within the confocal region is excluded by the pinholes, but fluorescence coming from more distant planes is accepted, thus limiting performance in very thick samples.
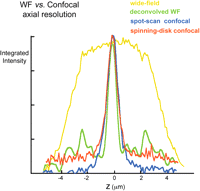
Axial resolution in wide-field (WF) and confocal microscopes. Three-dimensional stacks of images of a 0.2-µm diameter fluorescent bead were collected in a WF, disk-scanning, or spot-scanning confocal microscope. The graph shows the total integrated intensity from the bead in different focal planes, individually normalized for each microscope. The intensity profile in the WF images after deconvolution is also shown. The intensity recorded by the disk-scanning confocal microscope falls off exactly as with the spot-scanning microscope for small values of defocus but then plateaus and remains constant while the spot-scanning intensity continues to decrease to 0.
Troubleshooting Guide
This guide is primarily for confocal microscopes, but the same general principles apply to all methods. For troubleshooting deconvolution, see McNally et al. (1994, 1999) and particularly Wallace et al. (2001).
When poor images are obtained, the first question to be answered is whether the problem is with the specimen or with the equipment. An enormous amount of frustration and wasted time can be avoided if standard samples are available that can be used to compare the system performance at the moment with its performance in the past (i.e., on a day when good images were obtained). Four simple specimens are useful for this purpose: a resolution test target (e.g., see Fig. 29); an optically flat mirror or bare glass slide; 0.2–0.5-µm diameter beads labeled with multiple fluorophores (e.g., TetraSpeck; Molecular Probes/Invitrogen) that are excited by each of the laser lines and detected through each of the filter sets on the instrument; and a solution of fluorescent dyes covering similarly broad excitation and emission spectra (e.g., a mixture of 4′,6-diamidino-2-phenylindole (DAPI), Alexa Fluor 488, Alexa Fluor 594, and Alexa Fluor 633 solutions). The beads should be spread into a film on a coverslip, allowed to dry, and mounted on a thin layer of antifade solution or optical cement (e.g., Epo-Tek 301; Epoxy Technology). To make the fourth standard specimen, a generous layer of the solution of fluorescent dyes should be sealed under a coverslip.

Image of a resolution test target that was tilted by ∼1.5° on the microscope stage. The spacing of the gratings in micrometers is marked on the specimen. Because of the tilt, the defocus varies across the image as indicated by the orange scale. The specimen-to-lens distance increases from right to left. Note that the black–white bars of the 0.29-µm grating are fairly distinct at the left end but smeared to an average gray on the right with a 60× (NA = 1.4) objective, 546-nm illumination.
On a day when the equipment seems to be in good working order, collect and store a 2D image of the resolution test target at optimum focus, a 3D stack of images of the fluorescent beads, and an image of the fluorescent dye solution a few micrometers underneath the coverslip, using the smallest pinhole. Do this for all laser lines and all filter–detector channel combinations. Also collect an X–Z scan of beads as in Figures 28 and 30. For these measurements, place any gain, sensitivity, background, dark level, or other adjustments in a manual mode. Experiment to find settings of these parameters that give a zero intensity reading in the absence of illumination and a peak intensity reading that is just below saturation for the in-focus illuminated beads (see above section on Practical Aspects and Tips for Generating Reliable Images). Carefully record these settings along with the objective lens used and the parameters relevant to illumination and signal intensity (laser-tube current, neutral density filters, acousto-optic tunable filter settings, beam splitters, pixel spacing, pixel integration time, etc.). These measurements serve as a calibration that can be repeated later when the performance of the system becomes questionable. A list of the symptoms of some common diseases afflicting confocal microscope systems is given below.
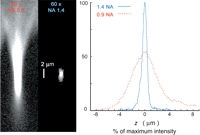
x–z scans of 0.9-µm-diameter fluorescent beads and measured axial intensity profiles. (Left) The left-hand side of the image shows a scan using a 40× (NA = 0.9) objective lens. The right half is a scan of a different sample using a 60× (NA = 1.4) lens. Intensity for the two images has been adjusted to the same maximum value; in reality, the 40× (NA = 0.9) image is much dimmer. The round bead appears to be much more elongated with the lower NA objective for two reasons. Decreasing the NA affects vertical resolution much more severely than lateral resolution. The left scan is also markedly asymmetrical above and below focus, indicative of spherical aberration, which causes further elongation. The right scan (of a different sample) also shows a small amount of spherical aberration. Scale bar, 2 µm. (Right) The graph shows the measured axial intensity profiles for these two situations. In these curves, spherical aberration manifests as asymmetry of the profile to the right and left of the peak (e.g., 40× curve beyond 5 µm).
Symptom: Image intensity is decreased over the whole field of view (at all magnifications).
Likely causes: If the image is at first bright but then gets dimmer, the problem is either photobleaching or drift of the focus level. Check for focus drift by collecting a reflectance image of a mirror or bare glass surface using the smallest possible pinhole. (This image is very sensitive to the focus level.) If the image is always dim, then there is probably a misalignment of the confocal pinhole or of the internal mirrors of the laser. If it is the laser, it will affect some wavelengths more strongly than others. For instance, the Ar/Kr and Kr lasers are very prone to loss of their yellow (568-nm) and red (647-nm) lines while retaining the blue lines (488 or 476 nm).
Symptom: Image intensity is decreased over the periphery of the field of view, which is more pronounced at lower magnification.
Likely causes: If the effect is seen in both fluorescence and reflectance images, then it is either a misalignment (most systems) or an intrinsic design problem (older Bio-Rad systems). If the effect is much more pronounced in fluorescent images than in reflectance images, then it indicates chromatic aberration.
Symptom: The resolution is poor.
Likely causes: If the problem is apparent both in thin (e.g., fluorescent beads) and thick samples, then the fault is probably in the alignment of the confocal optics (but first make sure the objective lens is clean). Check that the laser beam is correctly centered on the axis of the objective lens and that the entire back aperture of the lens is filled with incoming light. If the problem is restricted to thick samples, then spherical aberration is probably the culprit. The newer silicone-immersion and water-immersion objectives with a correction collar greatly ameliorate this problem with thick samples in aqueous media but at the cost of slightly decreased resolution with optically ideal specimens (i.e., very thin samples with a refractive index the same as glass and positioned immediately adjacent to the coverslip).
Symptom: Focusing for maximum brightness does not give the sharpest image. The image can be made bright or sharp but not both.
Likely causes: The system is not confocal. The focal plane for the illumination system does not coincide with the focal plane for the imaging system. For visible wavelength illumination and imaging, probably the pinhole or an intermediate lens that focuses the light on the pinhole is misaligned. If the illumination or imaging wavelengths are ultraviolet (UV) or far red, then a misaligned collimator lens is the likely culprit.
Symptom: Alternating stripes of higher and lower intensities appear in the image.
Likely causes: Mechanical vibration, defects on the scanning mirrors, or an electronic oscillation in the laser or the detector circuits are the likely causes. To decide between these, collect an image of the fluorescent dye test sample with the largest available pinhole aperture. This image is very insensitive to vibration but still sensitive to electronic oscillations and mirror defects. Mirror defects cause a fixed pattern of bright and dark stripes that does not change between images. Most electronic oscillations (and mechanical vibrations) give a different pattern with each image. Confocal systems from some manufacturers use single-mode polarization preserving fiber-optic coupling of the laser to the scan head. Some, but not all, of these systems are very prone to fluctuating illumination intensity and polarization angle. Painstaking rotational alignment of the fiber polarization axis with the laser polarization axis mitigates the effect, but within a few days, the fiber drifts out of alignment and the stripes reappear.
Symptom: A circular bright spot or a set of rings appears at a fixed point in every image.
Likely cause: A reflection of the laser beam off an internal glass surface is being detected. If fluorescence images are being collected, then an inappropriate set of filters is being used (reflected laser light is getting through). Most systems now include a quarter-wave plate and polarizer combination to minimize the problem in reflectance-mode imaging. Probably one of these elements has been rotated.
Symptom: Images from different fluorophores are misregistered.
Likely cause: Displacement of a short-wavelength (e.g., DAPI channel) or long-wavelength (e.g., CY5 or Alexa 633 channel) from the middle wavelength channels is usually because of misalignment of a UV–visible or infrared (IR)–visible collimator lens (a component of the illumination path that corrects for the small residual chromatic aberration present in all currently available objective lenses). Be forewarned: Some manufacturers’ service personnel do not routinely check collimator-lens alignment, so a recent inspection by unsupervised service personnel provides no assurance that the system is correctly aligned.
STRUCTURED ILLUMINATION METHODS
The goal of these techniques is to improve the images of thick objects by a combination of optical and computational manipulations. Normally one strives to achieve completely uniform illumination (i.e., completely “unstructured”) across the entire field of view, so that variations in intensity across the image arise solely from variations in the structure of the object. Contrary to what might be expected, superimposing artifactual intensity fluctuations across the image by using carefully patterned nonuniform illumination can actually increase the amount of information about the object that is stored in the image.
The development of new techniques for using structured illumination to enhance microscope performance is proceeding rapidly and in many directions (Bailey et al. 1993; Neil et al. 1997, 1998, 2000; Wilson et al. 1998; Gustafsson et al. 1999; Hanley et al. 1999, 2000; Gustafsson 2000; Cole et al. 2001; Heintzmann et al. 2001; Dubois et al. 2002). An excellent survey has been written for biologists by Heintzmann and Ficz (2007). As of this writing, most of the techniques have been shown only on very thin, fixed specimens and are available only in a few specialized laboratories. In this article, we describe one method (Neil et al. 1997, 2000) that does work quite well with living specimens of moderate thickness and is now commercially available from at least two vendors.
Optical Principles
The basic principle is shown in Figure 29, which illustrates, in a different way, the same information contained in Figures 1–3. The figure shows an image of a resolution test target, a series of gratings of different spacings, which was tilted by ∼1.5° before being photographed. The tilt, which is from right to left in the image, displaces the sample from the focal plane by an amount that increases, in opposite directions, away from the center of the field. Notice that the 0.33-µm grating is sharp and well resolved in the center but becomes quite blurred toward the left. The left side of the 0.29-µm grating is just resolved at a defocus of 0.25 µm, but the right side is completely smeared out when the defocus increases to 0.5 µm. How can one turn this “problem” into a “solution”?
Imagine the use of a grid of evenly spaced lines as a mask through which the specimen is illuminated (Fig. 31). The grid is placed in the light path of the microscope with appropriate lenses to bring its shadow on the specimen into sharp focus in exactly the focal plane of the objective lens. If the specimen is thin, then the result is an image of the in-focus specimen crossed by a set of sharply demarcated shadows where the illumination has been interrupted by the mask. If the specimen is thick, then superimposed on this in-focus image will be a blurred image of the other planes of the specimen (Figs. 5 and 31). However, the contribution from the out-of-focus planes of the specimen will not be modulated by the lines of the grating; away from the in-focus plane, the shadow of the grating quickly becomes smeared and the illumination is uniform, at a level that is the average of the light and dark bars. Note in Figure 29 that a defocus of 0.5 µm is sufficient to completely eliminate the 0.29-µm modulation.
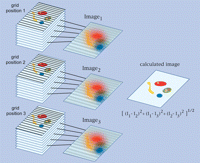
Structured illumination used for optical sectioning. The light source is masked by a regular grid, which casts a pattern of stripes on the in-focus object plane. Three images of the thick object are acquired, shifting the grid by one-third repeat between each acquisition. Each image is the sum of contributions from the in-focus plane, which is shadowed by sharply defined stripes, plus blurred out-of-focus planes without distinct stripes. The square root of the sum of the squared difference images removes the out-of-focus blurred light, giving the in-focus plane.
By a simple manipulation, the blurring caused by defocus can be turned into an effective tool for separating in-focus from
out-of-focus light when the light is right (i.e., structured). A simple algebraic combination of three images of the object
illuminated through a grating shifted by exactly one-third of its period between each image yields an in-focus image uncontaminated
by out-of-focus blur (Neil et al. 1997):
The simple sum of the three raw images (I1 + I2 + I3) is of course identical to the normal wide-field image with no grating. The best optical sectioning is obtained when the period of the grating is slightly larger than the diameter of the Airy disk (Neil et al. 1997).
The grating can be shifted rapidly and precisely (e.g., by a piezoelectric element), and the calculation is very fast. The rate-limiting step for optical sectioning by this method is thus acquiring the three images. Compared to wide-field plus deconvolution methods, the maximum acquisition rate would be threefold slower, but the finished product, the in-focus optical sections, is available immediately instead of after 1–2 min as for the deconvolution computation. The total exposure is slightly greater than for wide-field plus deconvolution methods because the bars of the grating are typically not completely opaque. The z-axis resolution is comparable to what is achieved by either confocal or deconvolution methods.
The limitations of this method are not fully known, but it seems that the method has trouble with structures whose sizes are comparable to the grid spacing. It is expected that the computation would also fail for extremely thick samples, when the background out-of-focus light becomes overwhelming. In that case, the contrast of the grid lines in the raw data will be very low, and the difference between I1, I2, and I3 would become comparable to the noise level. A discussion of some of the technical problems that need to be addressed to make the technique useful in practice has been published (Cole et al. 2001). The simplicity of the hardware component means that it is cheap to manufacture and can be user installed as an add-on to virtually any modern microscope. It is safe to predict that this and other variants of structured illumination microscopy will quickly supplant expensive confocal microscopes for many applications on modestly thick specimens, particularly living samples.
INTERPRETING THE RESULTS
The Meaning of Optical Section
Although the images produced by confocal microscopy, deconvolution, and structured illumination methods are referred to as optical sections, they differ from true sections in that their top and bottom edges are not sharply defined. In a physical section as cut by a knife in a microtome, there is no ambiguity about which section contains each point of the original object, at least not at the resolution of the LM. A specified point in the cell was either included in one particular microtome section or it was not, depending on its original position, but there is no intermediate state. An optical section, however, includes some locations fully (i.e., present at their true intensity) and other locations above and below at less than their true intensity. There is no sharp cutoff that demarcates what is included in the optical section and what is excluded. Instead, there is a continuous decrease in the ratio of image intensity to object intensity for locations further and further away from the midpoint (i.e., the CTF falls off steadily with the distance from the focal plane; Figs. 2 and 3).
A measurement that is commonly used as the analog of section thickness for optical sections is the full width at half-maximum (FWHM) of the curve that describes the relative intensity of points at different distances from the midpoint of the section. This curve can be measured by collecting a series of closely spaced optical sections (0.1 µm for the highest-NA lenses) of a small bright object. A small fluorescent bead is a good specimen for this measurement, but any bright object that is small compared with the expected FWHM can be used. A plot of the total intensity of the image of the object in each optical section should give a curve similar to the one in Figure 28, the axial intensity profile of the 3D PSF for the optical system (equivalent to a vertical line through the center of Fig. 3). For the highest-NA objective lenses, confocal, deconvolved wide-field images, or structured illumination microscopy should give a PSF having an axial FWHM of ≤0.6 µm. Much smaller values of FWHM have been obtained but not with samples and equipment that are realistic for live cell imaging experiments (Bailey et al. 1993; Hell et al. 1997; Gustafsson et al. 1999; Heintzmann and Ficz 2007).
Spherical Aberration
The FWHM of the vertical PSF decreases with the square of the NA of the objective lens. For confocal systems, the width also decreases with decreasing pinhole size. For all microscopes, the vertical PSF is very sensitive to the presence of spherical aberration (Figs. 9 and 30). Unfortunately, one is often forced to accept a certain amount of spherical aberration when examining living specimens (Fig. 24). The highest-NA oil-immersion objectives are designed for work with a specimen that is located immediately beneath a coverslip connected to the lens by immersion oil, which obviously cannot be true for all regions of a thick specimen. Consequently, the image is increasingly degraded by spherical aberration as the focal plane is set deeper and deeper into the specimen. The problem is greatly ameliorated by using water-immersion objectives. These are water-immersion objectives designed for use with a coverslip, in contrast to older designs (e.g., those used by electrophysiologists for patch-clamp studies). They perform better on thick specimens immersed in water than standard (very short working distance) high-NA oil-immersion objectives. However, the PSF of these water-immersion lenses is not yet as good as with the best high-NA oil objectives nor are the water-immersion lenses quite as bright. There are now objectives available designed for use with a silicone-immersion oil of refractive index ∼1.4 (see Fig. 9) with a correction collar that compensates for different depths of imaging.
As illustrated in Figures 9 and 30, spherical aberration leads to an unsymmetrical response to defocus; the image looks different when defocused by the same amount in opposite directions from the in-focus plane. When present, significant spherical aberration is readily visible. A convenient way to check for it is to find a very small, very bright dot of fluorescence and compare its appearance as the lens is defocused by a small amount (a few micrometers) in either direction. A typical observation is the appearance of bright rings on one side of focus and general fuzziness without rings on the other side. With the water-immersion lenses, this asymmetry can often be eliminated over an extended range of focal planes in a thick specimen by careful adjustment of the correction collar. With oil-immersion lenses, one can choose an immersion oil with a refractive index that minimizes the spherical aberration at the depth in the specimen where the major interesting features lie (for depths <∼15 µm), but optical performance in other focal planes will be degraded (Fig. 24).
Chromatic Aberration
With modern highly corrected objective lenses and an ideal sample, all wavelengths of light in the visible range should be focused to the same point at an accuracy of better than 0.1 µm (Keller 1995). However, if the mixture of refractive indices in the sample deviates from the design parameters of the objective lens sufficiently to cause noticeable spherical aberration, then chromatic aberration is also likely to be induced. The lens design is specific not only for a particular arrangement of refractive indices but also for the way in which those refractive indices vary with wavelength (dispersion). It is unlikely that the specimen dispersion matches the design specifications, resulting in a shift in focal position according to wavelength. Figure 32 shows this behavior in a series of optical sections of a single bead. In this case, the chromatic aberration is caused not by the sample but by misalignment of collimator lenses that are supposed to correct the confocal system for residual chromatic aberration in the objective, allowing it to be used with UV and IR as well as visible light.

Chromatic aberration in confocal images. Eleven optical sections, spaced at 0.2-µm intervals, of a single, 0.2-µm diameter bead labeled with four different fluorophores. The dotted cross in each section marks the x–y location of the center of the bead in the green image at best focus. Notice that the other three wavelength bands are shifted in x, y, and z with respect to the green. The yellow and blue images also reveal some spherical aberration. A composite color image for the sixth optical section is shown at the top. (Red) Excitation (ex), 633 nm; emission (em), >650 nm. (Yellow) ex, 568 nm; em, 585–615 nm. (Green) ex, 488 nm; em, 505 nm–600 nm. (Blue) ex, 364 nm; em, 385–470 nm with a 63× (NA = 1.2), water-immersion lens.
As the composite image at the top of Figure 32 shows, chromatic aberration is a serious problem in determining colocalization of different fluorescent molecules. In an extended sample, complicated artifactual shifts are observed because the direction and amount of apparent shift between different colors varies with position in the field of view. Interpretation of slight differences in localization demands careful control experiments to rule out chromatic aberration.
Chromatic aberration is of particular concern with confocal microscopes (Hell and Stelzer 1995; Keller 1995). If the focal spot for the illumination wavelength is not in the same position as the focal spot seen through the pinhole aperture, then the system is not confocal. In this situation, image intensity is severely decreased.
SNR
Live cell imaging is considerably more demanding than observation of fixed samples. The motivation to invest the extra time and effort is often the need to observe changes in distribution or amount of some fluorescent molecule. To be confident, however, that an observed change is real, rather than due simply to random fluctuations in the image intensity, the investigator must consider the SNR in the image. Some simple calculations are involved, but math phobes should not be discouraged. This simple calculation can prevent a lot of wasted time.
Suppose that an experiment requires detecting changes of 25% or more in the local concentration of a fluorescent molecule. As an example, consider the cells shown in Figure 7. The objective of the experiment will be to follow the incorporation and removal of YFP tubulin at the growing and shrinking ends of microtubules. Because this specimen is thin enough that the background from out-of-focus light is not overwhelming, it is appropriate to image it using any of the microscopic techniques described in this article.
To restate the experimental requirement, the SNR in the image must be large enough to be confident that a 25% increase or decrease in fluorescence at the end of a microtubule is not just because of random noise. To draw this conclusion with 95% confidence, the expected fractional change in signal (i.e., 0.25S) must be greater than twice the standard deviation (i.e., greater than twice the noise, 2N). Thus, SNR must be at least 8 for this measurement to succeed. Expressing the required precision as a fractional change P (i.e., 25% precision in the measurement of S means P = 0.25), then for 95% confidence, SNR must be >2/P.
To decide if this experiment is feasible, a rough estimate of the signal and its standard deviation is needed. First, locate
an area of a typical sample that is similar to the region of interest. Second, collect two images in rapid succession from
this area without changing anything between exposures (avoid photobleaching). In the absence of noise, these two images should
be identical. Thus, the difference between them can be used to estimate the noise level in a typical measurement. In fact,
the standard deviation of this difference image, call it s, is  times the standard deviation of a single pixel in the original images. The noise in the measurement of microtubule fluorescence
will be larger than s because the background will have to be subtracted. If the ratio of background to total intensity is b, then the standard deviation in one pixel of the background-corrected microtubule fluorescence image will be a factor
times the standard deviation of a single pixel in the original images. The noise in the measurement of microtubule fluorescence
will be larger than s because the background will have to be subtracted. If the ratio of background to total intensity is b, then the standard deviation in one pixel of the background-corrected microtubule fluorescence image will be a factor 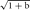 larger than the standard deviation in the raw-intensity measurement. In Figure 7, b = 0.95 before deconvolution and 0.8 after deconvolution. Finally, note that the target area for the measurement is more than
a single pixel. If the target includes n pixels and the average fluorescence per pixel of microtubule after background correction is denoted by F, then
larger than the standard deviation in the raw-intensity measurement. In Figure 7, b = 0.95 before deconvolution and 0.8 after deconvolution. Finally, note that the target area for the measurement is more than
a single pixel. If the target includes n pixels and the average fluorescence per pixel of microtubule after background correction is denoted by F, then
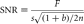
For the experiment in Figure 7, F was ∼50 (out of 4095 maximum for the 12-bit CCD), averaged over a 5 × 5-pixel box, s was ∼12, and b was 0.95, giving an SNR = 21. Thus, the images collected under these conditions are indeed good enough to detect the hoped-for change in YFP-tubulin incorporation.
Now suppose one contemplated doing this experiment with a spot-scanning confocal microscope instead of wide-field plus deconvolution microscopes. Doing this simple calculation beforehand would save a lot of time and frustration because one would probably find that the experiment cannot be performed with a spot-scanning confocal microscope. Typical numbers (Zeiss LSM510) would be roughly F ∼ 20 (out of 255 maximum for 8-bit detection), s ∼ 30, b = 0.5, which for the same size target gives a SNR = 4 (compare Fig. 7 and Fig. 22).
Point-scanning confocal microscope images are much noisier than wide-field images for several reasons. In a side-by-side comparison, using a very thin specimen with no fluorophores in out-of-focus planes, using the same lenses, same filters, same wavelength, and equal illumination doses, a typical wide-field microscope will collect two to four times more photons in the image than a spinning-disk confocal microscope. The efficiency of spot-scanning confocals is lower still; ∼50-fold fewer photons collected in the image than with a spinning-disk confocal microscope and several-hundred-fold fewer than a wide-field microscope, at an equal illumination dose. The lower quantum efficiency of the PMT detectors on spot-scanning confocals compared with the CCD detectors on spinning-disk or wide-field microscopes (∼15% vs. ∼70%) accounts for some of this lower efficiency but clearly not all.
The other reasons for these dramatic differences in efficiency are unclear at present, but they are observed in all machines tested and thus seem to be intrinsic properties of the different optical designs rather than examples of poor engineering in any one brand (Murray et al. 2007). In addition to having lower signals, many confocal microscopes add a large amount of unnecessary noise to the image, generated by electronic artifacts in the detector circuitry (Fig. 21) and random fluctuations in the laser illumination intensity. Figure 33 shows a direct comparison of the noise in the illumination of a laser-scanning confocal microscope and a wide-field microscope. It should be emphasized that this extra noise in the confocal microscope is entirely unnecessary, simply a matter of poor design in most of the commercially available instruments.
Fluctuations in illumination intensity in a confocal versus wide-field microscope. (Top) The average intensity in a 100 × 100-pixel image of a uniform, stable, fluorescent sample is plotted for a series of 1000 images acquired at 1-sec intervals, for a point-scanning confocal microscope and a wide-field microscope with a CCD detector. (Bottom) The intensity from a single pixel in a sequence of 100 successive images of the same specimen is plotted.
Disk-scanning confocals, which use CCD detectors, have much better SNR than point-scanning instruments for the same exposure levels. The noise level in images from the structured illumination method described above will be increased by the processing steps needed to calculate the in-focus image, but model calculations suggest that this effect will be less than a factor of 2. Thus, the structured illumination technique also has the potential for giving an image SNR substantially higher than the commercially available point-scanning confocals.
Why Confocal?
After this comparison of SNR with a thin sample, it is perhaps worth restating the message of the introduction to this article: Each of the microscopic techniques described here has its own realm within which it reigns unchallenged and outside of which it must take second place to other approaches. When specimen thickness is modest, the SNR of the raw data acquired with a wide-field microscope is high enough to allow excellent contrast enhancement by restoration methods such as deconvolution or structured illumination. As the thickness and background fluorescence increases, wide field and/or deconvolution eventually fail, and the best images will be acquired with a spinning-disk or array-scanning confocal microscope. Finally, when the sample is very thick, only point-scanning confocal or multiphoton techniques are useful. As a counterpoint to the thin-sample SNR comparison, Figure 25 shows a comparison between point-scanning confocal microscopes and wide-field plus deconvolution microscopes with a sample that is definitely in the confocal realm.
When a new specimen is to be imaged, it is convenient to have a rule of thumb for deciding which method of microscopy is likely to give the best results. The magnitude of a simple specimen-dependent parameter that we have called the haziness index (H) (Murray et al. 2007) provides a numerical criterion for choosing the most appropriate mode of microscopy. H is computed as a ratio of background to signal in a wide-field image: the intensity of background from out-of-focus fluorescence divided by the intensity of the fluorescence from a small in-focus object. Small here means comparable in size to the Airy disk. For specimens with very large background, the intensity of such small objects will be unmeasurable in a wide-field microscope, so it is necessary to estimate H by some indirect means. Often it will be possible to find a thin edge of the specimen where the background is not overwhelming and measurement of the intensity of some small object of interest is possible in a wide-field microscope. This number could then be combined with a wide-field measurement of the background in the thickest region of the specimen to give a good estimate of H. Alternatively, if both signal and background could be measured in a thin specimen, then knowing its thickness relative to a thicker specimen would be sufficient to calculate the value of H in the regions that are too thick for wide-field microscopy.
This sort of extrapolation will be imprecise, but fortunately all that is required is an order-of-magnitude estimate of H (Fig. 34). For specimens with H < ∼20, wide-field microscopy with deconvolution can provide the best images. Over the range 20 < H < 200, spinning-disk confocals are the best choice. From 200 < H < 1000, it will probably be necessary to use a spot-scanning confocal microscope, and beyond H = 1000, none of these methods is likely to be satisfactory.
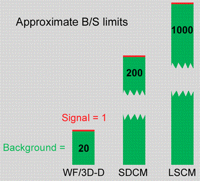
A rule-of-thumb guide for choosing, among wide-field and/or 3D-deconvolution microscopy (WF/3D-D), spinning-disk confocal microscopy (SDCM), and spot-scanning confocal microscopy (LSCM), the most appropriate mode of microscopy for a thick specimen. The magnitude of the ratio of out-of-focus background to the in-focus signal from a small object, which is a specimen-dependent parameter, determines the choice. For details, see Murray et al. (2007).
Three-Dimensional Reconstruction
The output from confocal microscopy, deconvolution, or structured illumination methods is typically a stack of optical sections, collected by changing the focus by a constant amount between each image acquisition. If the goal is to collect enough information to permit a 3D reconstruction of a specimen at the highest resolution possible, then adjacent optical sections will have to be spaced at increments of roughly half the FWHM of the vertical PSF. The resolution in the vertical direction will always be worse than in the horizontal direction, by approximately a factor of 3 at high NA. Thus, it is common in 3D reconstructions, particularly from confocal microscopes, for objects to appear elongated in the vertical direction (see Fig. 30). The software supplied for deconvolution often compensates for this effect, and a similar compensation (essentially a one-dimensional edge-sharpening filter) is helpful on confocal images.
Display of 3D image stacks in a form that efficiently and faithfully transmits the data to the viewer is a challenging task. Simplified representations of 3D data are essential for display purposes. Software abounds for 3D rendering or various other forms of displaying 3D-intensity distributions, and the output can be visually striking and quite persuasive. However, injudicious use of 3D-rendering software can also produce misleading representations that exaggerate the contrast (hence the reliability of the segmentation between different regions of the image) and the resolution (rendering fuzzy-intensity gradients as sharp boundaries). As always, caveat emptor.
- © 2011 Cold Spring Harbor Laboratory Press








